

Em alta


Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade

Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
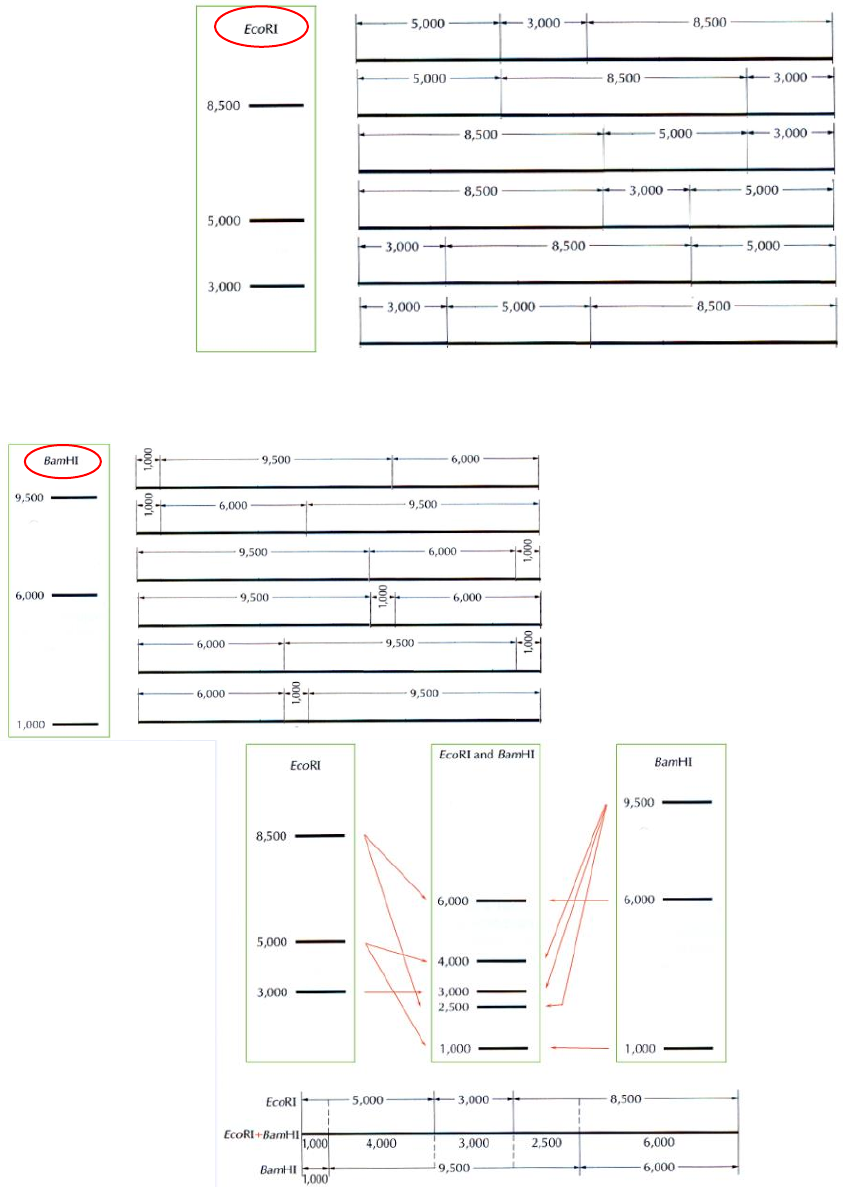
Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade

Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
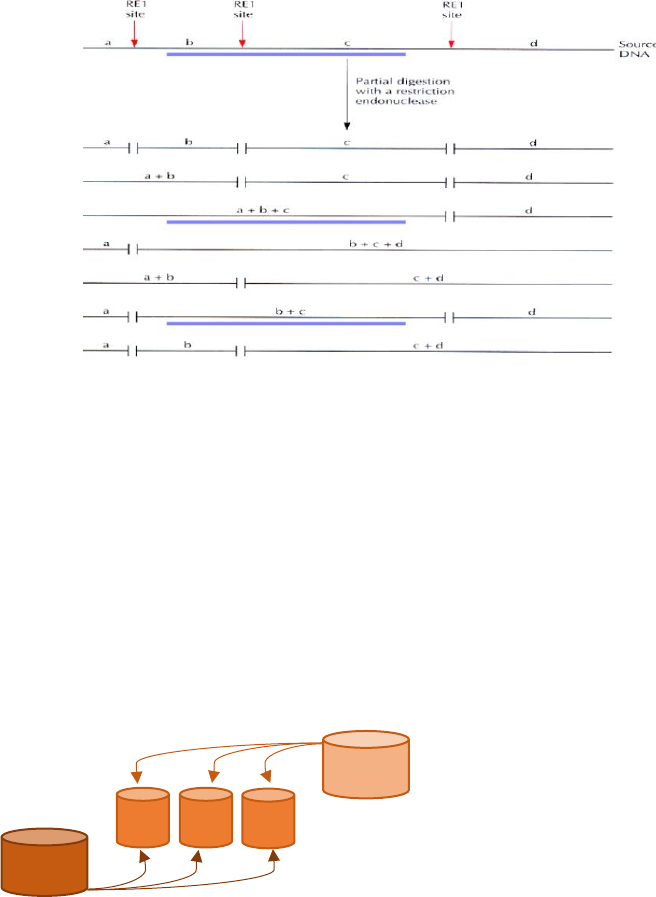
Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
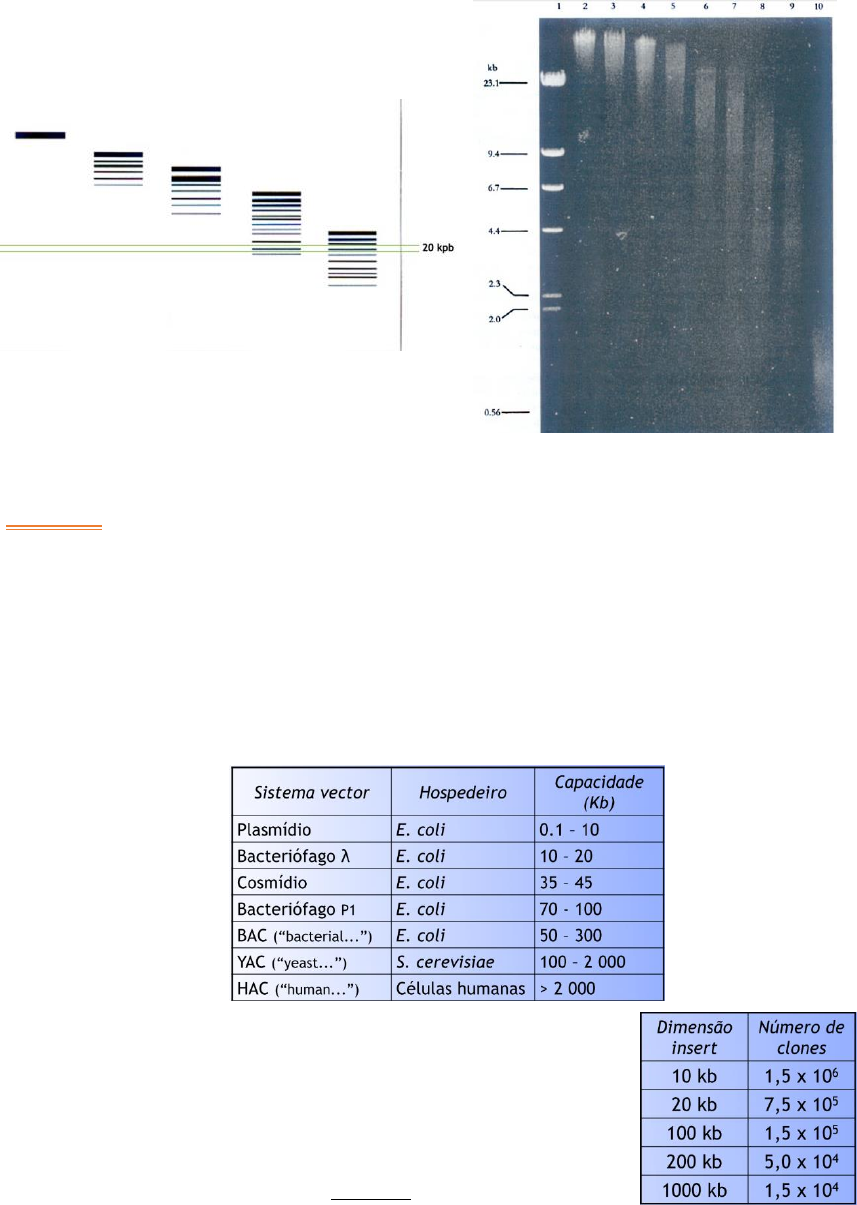
Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
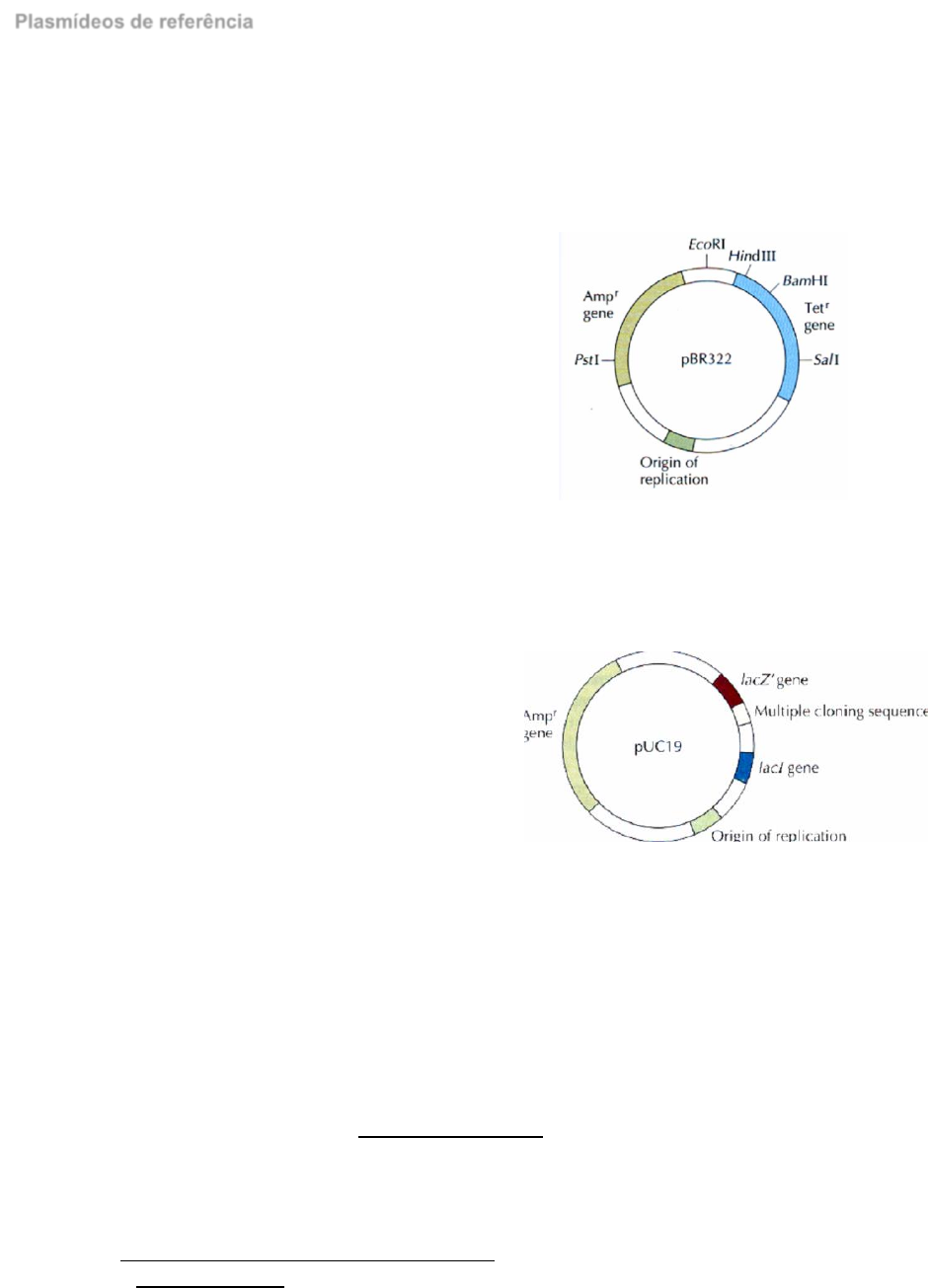
Crie sua conta grátis para liberar esse material. 🤩
Já tem uma conta?
Ao continuar, você aceita os Termos de Uso e Política de Privacidade
Prévia do material em texto
Engenharia Genética F2 1 I - CLONAGEM MOLECULAR A clonagem molecular é uma técnica de extrema importância a nível da Engenharia Genética que permite selecionar uma cópia de uma região específica do genoma e produzi-la em quantidades ilimitadas. As bactérias contêm plasmídeos, os quais podem ser usados como vetores de clonagem. Chamamos então vetor plasmídico a pequenas moléculas circulares de DNA derivadas de plasmídeos naturais de bactérias. Para proceder à clonagem: 1) Identificar DNA que nos interessa para o processo e extraí-lo do organismo dador. 2) Fragmentar o DNA que queremos clonar, e cortar os plasmídeos a usar como vetor, utilizando as mesmas enzimas de restrição, o que vai permitir compatibilidade de extremidades. As enzimas de restrição são endonucleases que tornam o DNA em cadeia simples, cujas extremidades têm cadeias específicas complementares com outras extremidades que tenham sido formadas pela mesma enzima. 3) Devido à especificidade do corte é possível o emparelhamento por complementaridade de bases entre o fragmento de DNA e o plasmídeo. 4) A ligação dos quatro extremos é feita por annealing e pela enzima DNA ligase, obtendo- se assim um novo plasmídeo recombinante. Annealing – as extremidades dos fragmentos de DNAs em cadeia simples unem- se numa ligação fraca por pontes de hidrogénio. A ligase – enzima que forma ligações fosfodiéster fortes entre os nucleótidos dos vários fragmentos. 5) O plasmídeo recombinante é introduzido numa bactéria hospedeira por transformação (ou conjugação ou transdução) que o vai encarar como DNA plasmídico normal e replica- o de igual forma como o resto do seu material genético, sendo assim possível criar milhões de cópias desse plasmídeo (por hereditariedade, ou seja, as células-filhas também o vão ter). 6) Selecionar os transformantes das células- filha que não ficaram com o plasmídeo por erros na replicação, e preservação e posterior utilização dos mesmos. Este processo não ocorre com 100% de sucesso, sendo isso, na verdade, bastante raro. É então necessário selecionar os plasmídeos que ficaram recombinados corretamente. Geralmente com o fragmento de DNA a clonar é colocado ainda no plasmídeo um gene que confira resistência a um antibiótico (por exemplo, a informação genética adicionada pode codificar uma proteína que degrada o antibiótico). Para selecionar os plasmídeos recombinantes basta submeter as bactérias ao dito antibiótico e selecionar aquelas que sobrevivem – significa que possuem o plasmídeo recombinante. Podemos então dizer que em clonagem molecular, a matéria-prima é composta por DNA insert, vetor e hospedeiro. Engenharia Genética F2 2 ENZIMAS DE RESTRIÇÃO Não são, como se possa pensar, um produto de laboratório, embora sejam de extrema importância em Engenharia Genética! As enzimas de restrição são endonucleases que existem nas bactérias como mecanismo de defesa que as protege de agressões de outros DNAs externos, impedindo que estes a transformem, sendo que cada bactéria tem a sua própria enzima de restrição. Estas atuam restringindo/ cortando os ácidos nucleicos infeciosos. Elas existem nas células sempre associadas a sistemas de modificação: enzimas de modificação – metilases – as quais colocam resíduos de metilo nos locais que são reconhecidos pelas enzimas de restrição no DNA celular para que este não seja afetado pelas enzimas de restrição erradamente, ou seja, protegem o DNA próprio da célula para que não seja destruído juntamente com o viral. G | AATTC As enzimas de restrição cortam sempre num local específico de uma sequência específica, que são próprios de cada enzima. Numa sequência de nucleótidos (sempre escrita na direção 5’3’) pode-se representar o local do corte feito pela enzima de restrição de várias maneiras. Vamos usar o exemplo da sequência utilizada acima, sendo que a enzima atua entre o nucleótido G e os dois As – sequência e locais SEMPRE reconhecidos pela enzima Eco RI. 5′𝐺↓𝐴𝐴𝑇𝑇𝐶3′ 𝐺:𝐴𝐴𝑇𝑇𝐶 𝐺|𝐴𝐴𝑇𝑇𝐶 𝐺𝑉𝐴𝐴𝑇𝑇𝐶 NOTA: As enzimas de restrição cortam sempre as duas cadeias da molécula de DNA, fazendo com que as extremidades fiquem em cadeias simples. NOMENCLATURA DAS ENZIMAS DE RESTRIÇÃO As enzimas de restrição são tipicamente identificadas por 3 letras que representam, respetivamente, o género, a espécie e a estirpe, e ainda 1 número que simboliza a ordem da descoberta. São exemplos: Hpa I / Hpa II: Haemophilus parainfluenza Eco RI / Eco RII: Escherichia coli R Local de atuação da enzima de restrição. Caso um dos outros ácidos nucleicos estivesse metilado, a enzima de restrição já não reconhecia o seu local e não atuava. Engenharia Genética F2 3 TIPOS DE ENDONUCLEASES DE RESTRIÇÃO Tipo I São complexas, multiméricas e combinam sistema de restrição e modificação, sendo que os mesmos complexos enzimáticos reconhecem uma sequência de DNA e modificam-na e cortam- na. Como não queremos as células modificadas não nos interessam nesta área. Cortam o DNA de forma random a partir da sequência de reconhecimento. Tipo II Estão são as enzimas de restrição utilizadas em laboratório, sendo as únicas tecnologicamente interessantes porque são pequenas, diversificadas e fáceis de manipular. Reconhecem sequências palindrómicas: sequências em que a complementar é igual à original, mas em sentido contrário, ou seja, como as cadeias são antiparalelas, lidas na mesma direção são iguais. Estas podem ser: Contíguas (GAATTC): ganham uma estrutura particular – estrutura cruciforme – a qual é reconhecida pelas enzimas de restrição. Não contínuas (GCCNNNNNNGGC): também adquirem a estrutura cruciforme mas não são úteis para a Engenharia Genética. Cortam em posições bem definidas, frequentemente dentro ou na periferia (muito próximo) da sequência de reconhecimento. Têm apenas atividade de endonuclease e não de modificação das células. Tipo III São grandes e combinam também restrição e modificação. Cortam fora da sequência de reconhecimento e raramente têm digestões completas. Não são interessantes para a Engenharia Genética. Tipo IV São grandes e combinam também restrição e modificação. Cortam fora das sequências de reconhecimento e estas são contíguas ou descontínuas. Não são interessantes para a Engenharia Genética. Engenharia Genética F2 4 TIPOS DE EXTREMIDADES OBTIDAS PELAS ENZIMAS As enzimas de restrição hidrolisam as ligações nucleotídica fosfodiéster entre o grupo fosfato e grupo hidroxilo, gerando uma extremidade 3’-OH e uma extremidade 5’-P. Se o local de clivagem não é no centro, a enzima gera extremos coesivos (“sticky ends”), que podem emparelhar com outros fragmentos digeridos pela mesma enzima. Estes extremos são mais fáceis de ligar pois a clivagem é assimétrica e ficam bases desemparelhadas – formam-se extremidades de cadeia simples. Se o local de clivagem é no centro, a enzima gera extremos cegos (“blunt ends”) pois a clivagem é simétrica. Não são interessantes na área da Engenharia Genética e Recombinação. Engenharia Genética F2 5 Enzimas correlacionadas Isosquizómeros: são enzimas que reconhecem a mesma sequência palindrómica mas uma gera extremos cegos e outra gera extremos coesivos que, por isso, não são compatíveis. Xma I (C|CCGGG) / Sma I (CCC|GGG) Hpa II (C|CGG) / Msp I (C|CGG ; C|C*GG) Isocaudómeros: são enzimas que reconhecem sequências palindrómicas diferentes mas geram extremos coesivos compatíveis que se ligam. Bam HI (G|GATCC) / Sal 3 AI (|GATC) O extremo compatível é GATC. ENZIMAS DE RESTRIÇÃO “MAIS ÚTEIS” As enzimas de restrição “mais úteis” são aquelas que… Reconhecem sequências palindrómicas. Reconhecem um nº de bases par – há enzimas que reconhecem nº de bases ímpar, mas não são interessantes na área da Engenharia Genética. As mais úteis são aquelas que reconhecem 4 ou 6 bases (é mais fácil encontrar sequências mais pequenas), pois as que reconhecem mais bases são muito específicas e só são aplicadas em processos em que conhecemos exatamente a sequência de nucleótidos. 4 bases: gera fragmentos estatísticos de 256 nucleótidos (nts). 6 bases: gera fragmentos estatísticos de 4096 nts. Geram extremos coesivos Reconhecem sempre a mesma sequência particular Têm uma grande capacidade de digerir o DNA Têm elevada pureza enzimática e elevada atividade específica. MAPAS DE RESTRIÇÃO Os mapas de restrição são uma compilação do número, ordem e distância entre os locais de corte de uma enzima de restrição ao longo de um segmento de DNA clonado. As unidades do mapa são expressas em pares de bases (ou para distâncias mais longas em pares de kilobases). Geralmente o mapeamento é a primeira etapa para caracterizar um DNA desconhecido. * Digestões simples: DNA digerido por apenas uma enzima de restrição. Faz-se uma determinação relativa das orientações dos fragmentos no DNA linear. Digestões múltiplas: DNA digerido por mais de uma enzima de restrição. Determinam-se as posições dos fragmentos de DNA produzidos pelas enzimas por eletroforese. Engenharia Genética F2 6 Quando se faz uma digestão simples apenas se sabe em quantos fragmentos de DNA surgem da digestão com um determinado enzima, o que corresponde com o nº de locais de atuação das enzimas de restrição. Contudo, há diversas hipóteses dos locais de corte. Se for um DNA circular e tiver apenas um local de corte, vai resultar num fragmento que é o plasmídeo original. Para saber os locais de corte exatos tem-se de fazer uma múltipla digestão (neste caso dupla). Com esta informação somos capazes de construir um mapa de restrição. Engenharia Genética F2 7 LIGAÇÃO DOS FRAGMENTOS Annealing: as extremidades dos fragmentos de DNAs em cadeia simples unem-se numa ligação fraca por pontes de hidrogénio. Ligase: enzima que forma ligações fosfodiéster fortes entre os nucleótidos dos vários fragmentos. OUTRAS ATIVIDADES ENZIMÁTICAS Fosfatase alcalina: enzima remove grupo fosfato da extremidade 5´ das moléculas de DNA. DNase I: enzima que degrada DNA em dupla cadeia por hidrolisação interna das ligações fosfodiéster. E. coli exonuclease III: enzima que remove nucleótidos dos extremos 3’-OH de moléculas de DNA. DNA INSERT O genoma de um organismo é demasiado extenso comparativamente com os genes isolados e, por isso, é necessário primeiramente saber o local de síntese da proteína de interesse (que pretendemos produzir), para que possamos obter o DNA do gene que a codifica. Vejamos então o exemplo da albumina humana (HSA). A albumina humana é sintetizada no fígado e depois é secretada. Vamos ter então de recolher células hepáticas e extrair-lhes ou o DNA; ou o mRNA (que representa menos de 1% do RNA total da célula) por identificação de caudas poli-A. Dentre este mRNA estará o mRNA percursor da albumina. A percentagem de DNA codificante é cerca de 1%, daí o mRNA ser 1%. Apenas o mRNA codificante das histonas é que não tem cadeias poli-A O mRNA extraído usando-se a técnica de cromatografia com oligo(dT). Os oligo(dT) têm uma sequência de Ts que reconhecem as caudas poli-A, emparelhando e capturando o mRNA, o qual é depois lavado e desnaturado para retirar da coluna (voltando depois a renaturar). Por fim, aplica-se a transcriptase reversa e dNTPs para obter o respetivo cDNA, usando como primer o oligo(dT) ou poli-U. Este cDNA é espontaneamente de cadeia simples, mas forma uma 2ª cadeia porque o RNA dobra-se sobre si mesmo. O cDNA já está processado (não possui intrões nem sequências de controlo da expressão) e por isso pode-se usar para clonar genes eucariotas em hospedeiros procariotas, pois a região codificante é contígua. No entanto, caso se use hospedeiros eucariotas, é melhor usar o gene nuclear em vez do cDNA. O cDNA não tem controlo de transcrição. Contudo, para ser clonado, o cDNA tem de ser linearizado primeiro. Em seguida junta-se linkers, ATP e a enzima ligase para que o cDNA fique com caudas de linkers, os quais são adaptadores sintetizados quimicamente, compostos por sequências palindrómicas reconhecidas por enzimas de restrição à nossa escolha. Após o tratamento com a enzima de restrição geram-se extremos coesivos e ficamos com o fragmento de DNA prontos a clonar. Engenharia Genética F2 8 Faz-se depois um screening dos clones de cDNA para pesquisar a sequência que nos interessa, o que se faz a partir de eletroforese, comparação das dimensões dos fragmentos, avaliação da atividade de enzima eventualmente expressa, hibridação molecular, etc.. Para isolar um gene de interesse, não se conhecendo o mapa de restrição começa-se por fazer uma digestão parcial do DNA com enzimas de restrição, de modo a obter as sequências de interesse. Esta digestão gera muito mais fragmentos do que a digestão completa, e além disso assegura que, caso haja local de corte no fragmento de interesse (como é o caso da albumina), conseguimos obter fragmentos inteiros, o que não seria possível com uma digestão completa (e por isso nunca se faz). No caso da albumina, o gene em que se insere tem 3 locais de corte por enzimas de restrição, incluindo um a meio do fragmento correspondente à albumina humana. Numa digestão completa, formaria 4 fragmentos distintos, cortando ainda o fragmento de interesse. Com digestão parcial consegue- se obter 7 possibilidades diferentes de corte, duas das quais não têm o fragmento da albumina cortado, sendo esses que se vão aproveitar. Para fazer uma digestão parcial tem-se: 1) Fazer dois tubos: um com uma certa quantidade de DNA e com enzima de restrição (10µg de DNA + 10 unidades de enzima de restrição); outro com a mesma quantidade de DNA e sem enzima de restrição (10µg de DNA + 0 unidades de enzima de restrição). a. Usa-se enzima de restrição de 6 bases que corta a cada ~4000 nucleótidos. b. O primeiro corresponde a 100% de digestão e o outro a 0% de digestão. 2) Fazer vários tubos com diferentes volumes dos tubos de (1), ficando assim com um gradiente de corte. Num destes tubos vamos conseguir obter o nosso fragmento de interesse inteiro. a. Poderia colocar-se o mesmo volume e incubar durante tempos diferentes, mas é um método mais trabalhoso e por isso geralmente não se faz. 3) Fazer eletroforese. Sabendo previamente o tamanho da sequência de interesse, através das bandas e dos marcadores de peso molecular conseguimos obter o nosso fragmento. a. Quanto mais tempo e maior for a concentração da enzima de restrição, mais fragmentos surgem e maior a sua separação no gel. A B C 100% 0% 1 2 3 3 2 1 Engenharia Genética F2 9 VETORES Os vetores são veículos possíveis de serem usados em clonagem, existindo imensas possibilidades de escolha. Para escolher o vetor mais apropriado deve-se ter em conta: O tipo de hospedeiro. O tamanho do DNA insert, uma vez que há vetores que suportam uma larga gama de tamanhos do mesmo, mas há uns têm uma capacidade mais reduzida e apenas suportam fragmentos mais pequenos. No quadro abaixo pode-se ver a comparação entre vários vetores e a sua capacidade (em kb) de receber o DNA insert. O número de clones (N) necessário obter, tendo em conta a dimensão do DNA insert, a dimensão total do genoma e a representatividade pretendida (f será a relação entre as dimensões), havendo fórmulas matemáticas que fazem esta estimativa entre a relaçãoe a probabilidade da existência de uma sequência (P), que é 1 em 1 milhão. 𝑁 = ln(1 − 𝑃) ln(1 − 𝑓) Engenharia Genética F2 10 TIPOS DE VETORES DE CLONAGEM Plasmídeos de referência Os plasmídeos são moléculas de DNA de cadeia dupla, circulares que existem em bactérias e no núcleo de alguns eucariotas. Replicam-se independentemente da célula. São os vetores mais importantes Têm dimensão variável entre alguns kb e 100kb (ou mais); e pode transportar até 10kb de DNA. O primeiro plasmídeo a surgir foi o pBR322, fabricado por dois mexicanos. Este possui origem de replicação de E. coli, dois genes de resistência a antibióticos (ampR para resistência à ampicilina e tetR para resistência à tetraciclina) e locais para reconhecimento específico por enzimas de restrição (EcoR I, BamH I e Pst l, Hind III e Sal I). O DNA insert é colocado no plasmídeo por substituição de um dos genes de resistência. Após a transformação da bactéria e reprodução, os hospedeiros são selecionados por resistência ao antibiótico, sendo as não resistentes aquelas que não têm o plasmídeo clonado. Das resistentes, apenas as que sobrevivem num meio com apenas um dos antibióticos e morrem em meios com o outro estão clonadas porque têm um gene de resistência substituído, uma vez que as que não morrem em nenhum têm o plasmídeo inteiro sem estar clonado. Atualmente já não se usa este plasmídeo porque não é viável para fragmentos maiores. Entretanto surgiu o pUC19 também com uma origem de replicação, mas com apenas um gene de resistência a antibióticos (o ampR) e com um gene de expressão da enzima β-galactosidase (lacZ), dentro do qual existe um MCS (com vários locais de corte reconhecidos por cerca de 20 enzimas, nomeadamente os que também haviam em pBR322). A seleção das bactérias neste caso é feita por resistência à ampicilina tal como no pBR322, mas das sobreviventes vai-se conseguir distinguir entre as que têm o gene de interesse e as outras consoante a coloração das suas colónias aquando do crescimento em meio com IPTG e x-gal. As colónias são então brancas se as bactérias tiverem o gene e azul caso contrário, sendo o azul a hidrólise do x-gal pela enzima β-galactosidase, com colaboração do IPTG. No caso de bactérias com DNA insert no MCS, vai haver uma inativação do gene produtor desta enzima, e assim não há hidrólise do meio e as colónias tornam-se brancas. Este é o plasmídeo mais usado. * Para ser um vetor de clonagem, o plasmídeo tem então de ter algumas características gerais: Uma região reconhecida como origem de replicação (ORI) pelo hospedeiro para que se possa multiplicar independentemente dos cromossomas bacterianos. Plasmídeos mais pequenos aproveitam as enzimas de replicação de DNA do hospedeiro, enquanto plasmídeos maiores podem transportar genes codificantes das suas próprias enzimas. Um gene que permita a seleção do hospedeiro (ex.: gene de resistência a antibiótico). Uma região polylinker ou local de clonagem múltipla (MCS – Multiple cloning site) reconhecida por enzimas de restrição à escolha, tendo de ser as mesmas usada para cortar o DNA insert, para que os extremos sejam compatíveis. Engenharia Genética F2 11 O MCS é um pequena fragmento de DNA que contém vários locais de restrição. Em vez de haver vários locais de restrição espalhados ao longo do plasmídeo, juntam-se todos num mesmo local. Os locais de corte correspondem a nucleótidos com sequências palindrómicas (que se lê de igual maneira de trás para a frente, e de frente para trás). Bacteriófago λ – Fagos São vírus que infetam especificamente as bactérias. É o mais usado depois do plasmídeo, e o mais usado pelos bancos celulares/genómicos (que trabalham geralmente com 20kb). O genoma tem dimensão de 49kb e pode aceitar até 25kb de DNA insert. O bacteriófago tem uma região do seu DNA codificante de uma proteína não utilizável que por isso pode ser substituída pelo DNA insert, o que é feito por recombinação de ambos os DNAs e reconstrução do vírus, com o apoio de enzimas de restrição e ligases. A sequência de DNA recombinado vai então ser repartida para integrar o bacteriófago – a sequência tem de ser cortada pelos locais COS (extremos coesivos) – e no final forma uma molécula de DNA circular. A parte substituída corresponde a qualquer uma que não seja necessária para sua replicação no laboratório. A substituição desta parte permite integrar DNA insert maior. Os locais COS permitem colocar uma grande molécula de DNA dentro da cabeça do fago, que de outra maneira não seria possível. O fago depois liga-se à membrana das bactérias e injeta o DNA recombinante no interior, sendo este replicado de forma independente ao genoma do hospedeiro, e recorrendo normalmente a enzimas codificadas pelo próprio DNA recombinante – ciclo lítico. O uso de vírus líticos (inativando a sequência de DNA que promove o ciclo lisogénico por integração no cromossoma da célula) são mais vantajosos porque permitem a libertação dos vírus da célula e sua propagação para as células vizinhas, tendo uma taxa de produção da nosso produto de interesse muito elevada; ao passo que um vírus lisogénico fica na fase latente em poucas células, e não se propaga. Cosmídeos São híbridos entre plasmídeo e bacteriófago – combinação de vetor plasmídico com local COS que permite a inserção de DNA na cabeça de fago . Têm uma elevada eficiência de transformação devido ao seu lado virião. Possibilita a inserção se fragmentos maiores, relativamente aos plasmídeos e aos bacteriófagos, podendo transportar até 45kb. Engenharia Genética F2 12 Primeiro este vetor vai comportar-se como plasmídeo: corta-se o DNA na zona do polylinker, dando origem ao genoma do fago, com local COS. O genoma fágico vai juntar-se aos fragmentos de DNA a clonar, formando cadeias concatenadas. Estas cadeias vão depois ser cortadas na zona COS para poderem ser integradas no bacteriófago. A sua principal vantagem é serem menos suscetíveis à degradação por nucleases do que o DNA de cadeia simples dos vírus. A inserção da extremidade COS vai permitir o circular do DNA. Os COS, embora se possam encontrar naturalmente em genomas de vírus e também aí circularizem o DNA, contudo os cosmídeos podem ser trabalhados em laboratório sem exigir manipulação de vírus. YACs – Yeast Artificial Chromosome São os menos usados A grande vantagem destes vetores é permitirem clonar sequências de DNA muito grandes, até 2000kb. As leveduras são organismos eucariotas que possuem cromossomas circulares, os quais se podem reproduzir em laboratório de modo a possuírem DNA que nos interesse produzir, sendo posteriormente multiplicados em leveduras (ou algumas bactérias). Os YACs são circulares e vão ter um local reconhecido por enzimas de restrição, formando assim dois braços (esquerdo e direito) em cujas extremidades se encontram telómeros para proteção do DNA linear da degradação por nucleases, e entre os quais é inserido o nosso DNA de interesse. No braço esquerdo vai ainda constar, além do telómero, o centrómero – local de ligação das fibras do fuso acromático que garante distribuição correta do cromossoma pelas células-filha durante a divisão celular –, 1 gene de resistência à ampicilina (ampR), uma origem de replicação de E.coli (porque é onde são construídos, manipulados e amplificados), uma ARS (sequência para replicação autónoma) e 1 marcador genético de auxotrofia para o triptofano (marcador de seleção metabólico). No braço direito vai ainda constar, além do telómero, 1 marcador genético de auxotrofia para o uracilo (marcador de seleção metabólico) Enquanto nos procariotas se acrescentam genes de resistência a antibióticos, nos eucariotas utilizam-se marcadores metabólicos de auxotrofia, permitindoa seleção de hospedeiros por crescimento em meios pobres na substância para a qual os marcadores são específicos (triptofano e uracilo). As leveduras que conseguirem crescer nestes meios são as transformadas com os YACs. Tem os elementos necessários para um cromossoma funcional Engenharia Genética F2 13 NOTA: Além destas características é ainda essencial que o vetor possua um local de terminação da transcrição. O gene de seleção por auxotrofia é essencial pois aquando da replicação o fuso acromático só se liga a uma certa quantidade de cromossomas. Ao adicionar o YAC, o numero de cromossomas torna-se superior à quantidade de fusos acromáticos e o nosso recombinante vai assim competir com os cromossomas da própria levedura. Desta forma vamos ficar com leveduras recombinadas e outras não, tendo-se de selecionar as de interesse com estratégias que permitam aumentar a estabilidade do nosso gene, tornando-o mais vantajoso, como é o caso da seleção por auxotrofia. LIGAÇÃO DO DNA INSERT AO VETOR Começa-se por usar a mesma enzima de restrição (ou isocaudómeros ou exonuclease para extremidades cegas) para preparar o DNA insert (obtido a partir do mRNA da proteína ou do gene nuclear) e o vetor de clonagem, como foi visto anteriormente, recorrendo-se a digestões parciais (não é necessário em DNA insert obtido por cDNA). Se em vez de extremos coesivos se gerarem extremos cegos, é necessário compatibilizar as extremidades usando linkers. O plasmídeo pode ter vários locais reconhecidos pelas enzimas de restrição, mas não queremos o DNA insert em todos eles, por isso não nos interessa uma digestão completa. Para obter um plasmídeo inteiro e linearizado é necessário uma digestão parcial, escolhendo-se depois o fragmento cortado no local onde interessa colocar o DNA insert. Após isto é preciso ter cuidado na ligação das extremidades, porque tanto os vetores como os DNAs podem voltar a fechar-se, devido à proximidade das suas extremidades, ou então unir-se de forma incorreta – há uma série de diferentes combinações possíveis mas apenas uma é do nosso interesse. Isto acontece porque se usa a enzima ligase para unir os extremos 3’-OH aos extremos 5’-P, num processo dependente de ATP. Contudo, ela não sabe quais os extremos do plasmídeo e quais os do DNA insert. Para impedir que o vetor feche recorre-se à enzima fosfatase que desfosforila os extremos 5’ do plasmídeo (a ligase não vai unir dois extremos OH), permitindo assim que este ligue apenas a DNA insert, embora uma das cadeias, como não tem P, não vai ficar ligada – nicks. Este método não impede no entanto a ligação entre inserts e sobre eles próprios, embora essa situação não seja tão grave porque não se conseguem replicar. As ligações corretas entre vetor e DNA insert vão depois ser colocadas no hospedeiro, e este deteta os nicks e repara-os. Engenharia Genética F2 14 TRANSFORMAÇÃO DOS HOSPEDEIROS EXPERIÊNCIA DE GRIFFITH’S (1928) Nesta experiência estudou-se a bactéria Streptococus pneumoniae que provoca pneumonia no ser humano e é, geralmente, letal nos ratos. Para tal foram usadas duas estirpes, com diferentes graus de virulência. A estirpe S, de virulência normal, é coberta por uma cápsula polissacarídea, o que confere uma aparência lisa às células. Se forem injetadas nos ratos, estes contraem pneumonia e acabam por morrer. A estirpe R, um tipo mutante não virulento, que não é coberta por cápsula, o que torna o seu aspeto rugoso. Se forem injetadas nos ratos, estes sobrevivem e permanecem saudáveis As células não são letaisr para os ratos. Fervendo as células S elimina-se o seu DNA (“morrem”) e resta apenas a cápsula da bactéria. Se esta for injetada nos ratos, eles sobrevivem e permanecem saudáveis, concluindo assim que não é a cápsula que é patogénica. Contudo, adicionando-lhe novamente DNA, a bactéria volta a ser patogénica. Injetou-se então uma mistura de células S (mortas pelo calor) e células R (vivas) nos ratos, os quais contraíram a doença e morreram. Foram ainda encontradas células vivas do tipo S no sangue dos ratos mortos. Isto é explicado pelo facto de células R adquirirem a cápsula das células S, apesar destas estarem mortas – Houve transformação das células mortas S pelo conteúdo das células R, o que vai alterar o fenótipo da bactéria, uma vez que esta ganha genes codificantes da cápsula, o que a torna patogénica. O Princípio da Transformação Genética diz que bactérias com fenótipo alterado terão o seu genótipo também alterado. MECANISMOS DE TRANSFORMAÇÃO NATURAL O DNA é físico-quimicamente estável e por isso dificilmente se quebra em fragmentos pequenos, o que representa uma dificuldade acrescida na sua incorporação pelas células uma vez que a parede bacteriana não permite a passagem do DNA inteiro. Desta forma, o material genético vai ter de entrar na célula por transporte ativo como se fosse para alimentação, ou seja, através de orifícios perto dos flagelos ou cílios. O DNA é então assim incorporado e, sendo estes locais de grande ocorrência de exonucleases, degradado. Contudo, pelas exonucleases 3’ e 5’ terem diferentes velocidades de atuação e Engenharia Genética F2 15 dependendo da disponibilidade de cada uma das extremidades, vai haver uma das cadeias que é mais rapidamente degradada do que outra. A cadeia simples mais lentamente degradada tem assim possibilidade de chegar perto do DNA celular e incorporar o genoma da bactéria. Este é um procedimento extremamente raro, que em termos estatísticos é nulo, mas que na prática ocorre. Transformação com DNA livre: quando uma bactéria morre e liberta o seu DNA, este pode ser incorporado por outras bactérias envolventes, caso o recombinante lhe permita adquirir características vantajosas em termos de sobrevivência (é o que acontece com as bactérias hospitalares). Transformação com plasmídeo: o plasmídeo é captado pela bactéria, havendo transformação da mesma. É um processo que ocorre pouco mas quando ocorre, o DNA não é degradado pelas exonucleases, uma vez que estas não conseguem atuar em DNA circular. Isto garante uma maior estabilidade ao material genético, embora dificulte de certa forma a sua transmissão. É esta que se usa em laboratório. Transdução – utilização de vírus: transferência de DNA de uma célula para outra através de um vetor viral (vírus bacteriófago). Os vírus são compostos de proteínas (que constituem a sua cápsula) e ácidos nucleicos, mas não têm organelos celulares que lhes permitam a sua reprodução, pelo que para realizar esse processo, é necessário a sua entrada em bactérias e o uso do teu seu ATP e organelos. Existem 3 métodos de entrada do DNA (ou RNA) viral nas bactérias: 1) Injeção do material genético (modo direto) Apenas o material genético do vírus é fundido na membrana celular, permanecendo a parte proteica no lado externo. 2) Fusão do envelope viral Funde-se com a membrana celular e o genoma do parasita invade a célula. 3) Endocitose O vírus consegue “enganar” os recetores químicos da membrana celular, que vão promover a fixação do vírus, que é englobado por invaginações da membrana. Engenharia Genética F2 16 Uma vez dentro da bactéria, o DNA viral pode prosseguir duas vias: Ciclo lítico: as funções normais da bactéria são interrompidas na presença do material genético viral, que prossegue imediatamente para replicação. Durante a replicação ocorre simultaneamente a síntese de proteínas suas que constituirão a cápsula que vai envolver os ácidos nucleicos, formando novos vírus. Isto acaba por provocar lise celular e libertação de inúmeros vírus funcionais, que vão, por sua vez, atacar outras células. Ciclo lisogénico: combina-se com o DNA da célula hospedeira e vai ser replicado juntamente com o genoma celular– fase dormente (não produz viriões). Nesta fase a sequência de DNA viral está reprimida e é como se nem lá estivesse. Se a bactéria for exposta a um fator que diminua a ação do repressor, o DNA viral deixa de estar “adormecido” e o DNA da células hospedeira começa a tentar reparar-se, expulsando o DNA viral. O vírus fica então ativo e prossegue para o ciclo lítico. A transdução pode então basear-se em ambos os ciclos, classificando-se em transdução generalizada e transdução específica: A generalizada baseia-se no ciclo lítico, durante o qual pode haver incorporação de DNA bacteriano aleatório nos novos vírus formados, o qual vai depois infetar outra bactéria. Aqui, o DNA da bactéria “doadora” incorpora o DNA da bactéria “recetora” (transduzida), sendo replicado juntamente com ele. A específica baseia-se no ciclo lisogénico, no final do qual a bactéria segue o caminho lítico. Quando isso acontece o DNA bacteriano (no qual está incorporado o viral) é cortado para formar novos vírus. Caso este corte seja defeituoso, juntamente com o fragmento viral poderá vir um pequeno fragmento de DNA bacteriano, que vai ser incorporado no vírus e libertado, podendo infetar outras bactérias, que ficam transduzidas, tal como na generalizada. Todavia, neste caso, os genes transferidos só podem ser aqueles imediatamente adjacentes ao fragmento de DNA viral. Engenharia Genética F2 17 Geralmente o DNA viral é injetado na bactéria (método direto), e o que se pretende é que o vírus seja não lítico e que integre o seu DNA no genoma da célula, ou seja, que siga o ciclo lisogénico e não destrua as células. O vírus lisogénico não é muito rentável em laboratório. A desvantagem deste método geral da transdução é a possibilidade de os vírus serem patogénicos. Conjugação (F+ e Hfr): transferência de material genético entre duas células, organismos ou bactérias, envolvendo o contacto entre elas mas continuando ambos os organismos a existir separadamente – ocorre com muita frequência. Este fenômeno foi descoberto através de duas variedades geneticamente diferentes da bactéria E. coli, que foram cultivadas juntas. Uma das bactérias doa o DNA e a outra recebe-o. A capacidade de doar DNA está ligada à presença do plasmídeo F (de fertilidade): as bactérias F+ são doadoras e as F- são recetoras. 1) Forma-se o pilus, que une as células e se vai retraindo até surgir um “canal de interligação” entre elas O par de células fica estabilizado. 2) O plasmídeo F é separado em dois, e uma das partes migra para a célula F-. A parte que fica na célula F+ é automaticamente replicada. 3) Ocorre replicação da parte que fica na célula F-. 4) Completa-se a transferência de DNA e as células separam-se. A doação de DNA faz-se então por troca de plasmídeos. Contudo, a incorporação do fator F pelo genoma da bactéria não é controlada tecnologicamente, acabando por não ser muito útil. Por vezes, uma pequena parte do DNA cromossómico une-se ao plasmídeo e é também transferido, podendo sofrer recombinação com o cromossoma da bactéria recetora. Isto aumenta a variabilidade genética da população bacteriana e também permite adaptação ao meio. As bactérias que possuem os plasmídeos recombinados chamam-se Hfr (High Frequency of Recombination). Não é adequada para uso tecnológico. NOTA: Juntamente com o gene F, passa-se o gene R que é o de interesse tecnológico, o qual confere uma característica vantajosa em termos evolutivos, como por exemplo resistência a antibiótico Engenharia Genética F2 18 Transposão: transferência de genes de uns cromossomas para outros, resultando na inibição ou ativação de outros genes – movimentação de partes móveis de DNA de uma região do genoma para outra. Do ponto de vista tecnológico são usados, mas têm a dificuldade de nunca se saber onde vão ser incorporados, acabando por não interessar muito (não se usam). Podem resultar em doença ou variabilidade genética. Permite que o ambiente de expressão mude. Inicialmente temos um plasmídeo que contém o transposão (Tn), que corresponde ao gene “amovível”, e o cromossoma-alvo, ambos com estrutura circular. Estes vão sofrer um corte por enzima de restrição e tornar-se lineares, e simultaneamente vai haver duplicação do Tn, e o gene fica com um transposão em cada extremidade. Após isto haverá cointegração do gene-dador no cromossoma-alvo, adquirindo o recombinante no final estrutura circular. Durante este processo há disrupção dum gene no cromossoma-alvo no meio do qual é colocado o gene com os transposões. Os transposões vão depois emparelhar e recombinar, o que leva à libertação de ambos os plasmídeos, ficando um transposão em cada. No final vamos então ter o transposão inicial e cromossoma-alvo com o transposão inserido. MECANISMOS DE TRANSFORMAÇÃO ARTIFICIAL Em células bacterianas (como a E. coli) geralmente recorre-se a transformação artificial, para preparar bactérias competentes. Contudo, este processo é bastante improvável de acontecer devido à ação das enzimas de restrição que impedem a transformação das bactérias. Para aumentar a probabilidade podem ser feitos dois tratamentos: Tratamentos químicos com cálcio, manganésio, etc., nos quais se exausta energeticamente as bactérias, tornando-as competentes para receber o DNA (as bactérias são modificadas), após a inserção do qual se fornecem condições às bactérias para elas recuperarem. O Ca e o Mn constituem pequenas moléculas que entram passivamente nas células, tendo estas de os excretar por transporte ativo usando mecanismos de iões bivalentes. Se administrarmos grandes concentrações destas moléculas, a bactéria fica esgotada energeticamente. A administração de Ca aumenta a eficiência de recombinação em 106 (ou seja, por cada µg de DNA tem-se 106 recombinantes), e se este for associado ao Mn, a eficiência sobe para os 108. Engenharia Genética F2 19 Tratamentos físicos como a eletroporação, processo no qual se submete as bactérias a uma corrente elétrica que altera a estrutura celular sem destruir o organismo, abrindo orifícios que permitem a passagem do material genético. Para tal é necessário uma corrente rápida e potente, tendo de se controlar bem estes parâmetros (se o tempo for demasiado pequeno, os orifícios fecham demasiado rápido). Este é o tratamento de eleição, embora seja dispendioso. Aumenta a eficiência de transformação para 1010. Transdução: não sendo propriamente um tratamento para aumentar a eficiência de transformação, pode constituir um método de transformação artificial. Em células eucariotas também se usam mecanismos de transformação artificial, tais como: Gene gun: dispositivo que dispara DNA a alta velocidade e induz a entrada física deste na célula. Já não se usa. Microinjeção; Eletroporação; Infeção viral * Os elementos genéticos passíveis de ser usados na transformação artificial são os plasmídeos (replicação independente), os episomas (livres ou integrados) e os transposões (integrados inespecificamente) e vírus (com ciclo parcialmente extracelular). Em todo o caso, o DNA que queremos transformar tem de apresentar uma vantagem seletiva que no caso das bactérias consiste na resistência a um antibiótico, enquanto nas células eucariotas trata-se de uma vantagem metabólica (como foi visto nos YACs). SELEÇÃO DOS HOSPEDEIROS RECOMBINANTES Já vimos atrás como funciona a seleção dos hospedeiros recombinantes para o caso dos plasmídeos pBR322 e pUC19. Também já vimos que, juntamente com o plasmídeo recombinado (com insert) que nos interessa, podem surgir outras moléculas como: 1) Plasmídeo com insert invertido (insert colocado ao contrário no plasmídeo que, mesmo sendo recombinante, não vai exprimir o produtodesejado), com fragmento do insert. com múltiplos inserts ou com fragmentos contaminantes. No caso de pUC19, nenhuma destas moléculas vai exprimir a β-galactosidade, mas também não têm interesse. 2) Plasmídeo fechado sobre si mesmo, direto ou invertido. No caso de pUC19, estes vão exprimir a β-galactosidade, sendo descartados. 3) Plasmídeo recircularizado que consiste num plasmídeo mutado No caso do pUC19 não exprime a β-galactosidade. 4) Fragmentos diversos recombinados ou não sem plasmídeo – são irrelevantes. A eficiência de transformação é calculada pelo nº de recombinantes a dividir pelas µg de DNA. Engenharia Genética F2 20 Em pUC19, o caso 4) é eliminado pelo antibiótico e o caso 2) é eliminado aquando do teste com o meio IPTG+x-gal (substrato cromogénico) porque forma colónias azuis, sendo automaticamente descartados. O problema serão todos os plasmídeos recombinados que não exprimem a β-galactosidade, tendo de selecionar-se entre eles, o do nosso interesse (com o insert direto). Para tal recorre-se uma seleção específica. Resumindo, temos portanto uma seleção em duas fases: 1) Uso de marcadores genéticos com possibilidades transformantes, tais como: substratos cromogénicos (IPTG/x-gal), inativação por inserção (β-galactosidase) e complementação de mutações definidas. 2) Seleção específica por hibridação com sondas moleculares de ácidos nucleicos ou por seleção imunológica (baseada na expressão das proteínas) Como teste de confirmação, no final, podem ser usadas várias tecnologias de análise dos genes clonados como: tradução in vitro de mRNAs, mapas de restrição, técnicas de blotting (Southern, Northern, Western, Dot-blot) e sequenciação. A última corresponde à forma mais fidedigna de conhecer o recombinante. HIBRIDAÇÃO MOLECULAR: uso de sondas nucleotídicas com o seu DNA marcado radioactivamente (caiu em desuso por ser prejudicial à saúde) ou por luminescência, o qual hibrida com as bactérias transformadas e liga-se por complementaridade ao insert, sendo este o processo mais usado. As sondas usadas consistem: No próprio DNA do insert, o qual é marcado e desnaturado, ficando em cadeia simples, e assim consegue hibridar com a bactéria. Num plasmídeo pré-existente específico que pode ser um fragmento conhecido ou uma sequência correspondente a regiões conservadas entre proteínas de várias espécies. No DNA total do dador do insert, o que só é possível se o DNA não for do próprio organismo. cDNA do dador do insert, que tem um tamanho menor do que o DNA total e vai fazer um screening apenas dos RNAs em expressão. Oligonucleótidos artificiais ou fragmentos de PCR, mas só podem ser usados conhecendo a sequência do insert. Os oligonucleótidos são difíceis de desenhar por se basearem no código genético que é degenerado, sendo feitas com base na sequência de aminoácidos da proteína codificada pelo gene-alvo: se não soubermos ao certo o tripleto (a sequência de DNA complementar ao gene-alvo) temos de pôr todas as combinações de aminoácidos possíveis (pois um aminoácido pode ser codificado por vários tripletos) – vamos ficar com muitas (demasiadas) sequências para combinar com o insert. A marcação das sondas, por sua vez, pode ser feita de três maneiras: Random printing: é a técnica mais usada e consiste em desnaturar por calor o DNA da sonda. Em seguida são adicionados oligonucleótidos que hibridam em ambas as cadeias e funcionam como primers. Por ultimo junta-se DNA polimerase I (Klenow, ou seja, sem atividade de exonuclease), 3 dNTPs normais e 1 marcado. Vão então formar-se cadeias em que um nucleótido está marcado e pode ser detetado por Southern blotting. Engenharia Genética F2 21 Nick-translation: é semelhante ao random printing, mas aos poucos está a ser substituída pelo mesmo. Nesta técnica submete-se o DNA da sonda à ação da DNase que promove a formação de uma abertura em cada uma das cadeias (em extremos opostos). O DNA é depois incubado com DNA polimerase I (que além de polimerase 5’3’, tem também ação de exonuclease) e dNTPs radioativos. Assim a polimerase liga-se à extremidade 3’-OH proporcionada pelo primer e vai remover a restante cadeia de DNA original, e sintetizar uma nova cadeia (complementar à adjacente) com os nucleótidos radioativos disponíveis. Marcação terminal: é uma técnica que já não se usa, mas que consiste na marcação num dos extremos (3’ ou 5’) com 32P radioativo que permite a deteção da sonda. SELEÇÃO IMUNOLÓGICA Deteção da proteína recombinante: faz-se um screening da proteína por imunodeteção (western blotting). Deteção da atividade proteica: verifica-se se a proteína está a desempenhar as duas funções, ou seja, se há reação enzimática. Identificação de fenótipo: consiste em observar as características fenotípicas (metabólicas) resultantes da expressão da proteína codificada pelo insert por complementação funcional. MAPAS DE RESTRIÇÃO: conhecendo o mapa de restrição do DNA insert para determinada enzima, podemos fazer uma digestão parcial e assim descobrir se na amostra usada existem fragmentos com tamanho idênticos ao insert. Pode assim, ser útil na seleção específica de recombinantes. BLOTTING Southern blotting Após uma eletroforese em gel de agarose para separar os fragmentos de DNA, para que se consiga ligar uma sonda ao DNA-alvo, este deve estar em cadeia simples, o que é conseguido por desnaturação. Contudo, o gel da eletroforese derrete com o aumento da temperatura, e por isso é necessário primeiro transferir o DNA para uma membrana, para que se possa aplicar a sonda. Isto é feito da seguinte forma: Contudo, a proteína pode apresentar mutações ou folding errado, e por isso estes métodos não são muito usados, embora nem todos os clones sejam iguais e possa haver proteínas com folding adequado entre as outras. A baixa frequência de clones selecionáveis torna o método pouco vantajoso. Engenharia Genética F2 22 1) Põe-se uma folha de nylon ou microcelulose por cima do gel de eletroforese. 2) A folha de nylon é coberta por muito papel absorvente e um peso no topo (0,5kg). 3) Estas camadas são todas colocadas em cima de papel absorvente em forma de ponte, cujas pontas são mergulhadas numa solução alcalina. Este procedimento vai permitir a migração do gel para a folha de nylon (de nitrocelulose) por capilaridade – o papel absorvente “puxa” o líquido e arrasta os fragmentos de DNA, que ficam na folha de nylon – Transferência por Southern (por isso se dá o nome de Hibridação de Southern). Para que os fragmentos de DNA fiquem bem fixos à membrana de nylon, submete-se a mesma a uma temperatura de 120oC durante 30min ou a luz UV. Em seguida: A temperatura elevada vai permitir a ligação química entre a sonda e o gene-alvo. Para depois estudar o gene em causa é necessário realizar nova eletroforese para isola-lo. * Hibridação em colónias: pode-se usar uma técnica semelhante ao Southern blotting em colónias, mas no qual não é necessário recorrer à eletroforese, pois a técnica é aplicada in situ. Assim, as células são transferidas para uma membrana (equivalente à folha de nylon) e depois provoca-se a lise celular e a desnaturação do DNA, bem como a sua ligação à membrana. O DNA é depois hibridado in situ com a sonda, que se liga apenas ao DNA de interesse, identificando-o. Northern blotting Processo muito semelhante ao Southern blotting, mas no qual se usa RNA em vez de DNA. Contudo, é preciso ter muito cuidado quando o RNA é tratado. 1) Eletroforese de RNA (o RNA também tem carga negativa). 2) Fragmentos de RNA tratados com formaldeído para desnaturar. 3) Transferência para membrana – Northern. 4) Hibridação com sonda. Exemplo de aplicação: quando queremos saber onde está o mRNA e uma extração total de RNA. Coloca-sea folha de nylon num saco com solução de hibridação e a sonda, fechamo-lo e colocamo-lo em banho de água a 60oC. Enrolamos a folha de nylon num fraco cilíndrico com a solução de hibridação e a sonda, que vai a um forno que roda. OU Engenharia Genética F2 23 Western blotting ou immunoblotting Para detetar uma proteína particular numa mistura utilizam-se anticorpos como sondas. Começa- se por desnaturar as proteínas com SDS e depois aplica-las a um gel de eletroforese de poliacrilamida. Para poder aplicar os anticorpos tem-se de transferir as proteínas para uma membrana, mas como a poliacrilamida torna o gel mais compacto do que a agarose, esta transferência tem de ser feita por transferência elétrica, aplicando uma corrente elétrica. Após isto os anticorpos são administrados (podem ser radioativos) que se vão ligar especificamente aos antigénios, revelando qual nossa proteína de interesse. * Seleção por expressão: pode-se usar uma técnica semelhante ao Western blotting em colónicas, mas no qual não é necessário recorrer eletroforese, pois a técnica é aplica in situ. Assim, as células são transferidas para uma membrana (equivalente à folha de nylon) e depois provoca-se a lise celular e a ligação das proteínas à membrana. A membrana é em seguida tratada com os primeiros anticorpos, depois lava-se (para remover aqueles que não ficaram ligados) e é tratada com segundos anticorpos, lavando-se novamente no final (para remover os 2os anticorpos que não ficaram ligados). Com a ligação dos anticorpos é possível fazer uma seleção in situ dos recombinantes. Engenharia Genética F2 24 Engenharia Genética F2 25 II - MANIPULAÇÃO DE ORGANISMOS MANIPULAÇÃO DE PROCARIOTAS Para a expressão de um gene clonado num procariota é necessário um conjunto de elementos: Elementos para transcrição (TATA box, promotor e terminador da transcrição) Sequências para a tradução (RBS (sequência de Shine-Dalgarno), AUG e codão STOP) Elementos para processamento (péptido de sinal para secreção da proteína final se for necessário) Não hidrólise do produto – o normal é o organismo degradar a nossa proteína de interesse porque ela é-lhe estranha. Para garantir uma maior estabilidade da proteína final e diminuir a sua degradação usa-se por exemplo uma proteína de fusão, ou seja, à nossa proteína de interesse é acoplada de uma proteína própria do organismo hospedeiro. Função biológica do produto pode não ser compatível com o organismo hospedeiro e por isso é necessário regular a sua expressão (por exemplo: clonar enzimas de restrição de outra bactéria em E. coli pode resultar na degradação do DNA desta). Nos procariotas produz-se um mRNA mais simples (relativamente aos eucariotas) e não ocorre a sua maturação, ou seja, quando ele é transcrito pode ser logo traduzido. A transcrição e a tradução ocorrem no mesmo compartimento, pelo que os processos acabam por ocorrer em simultâneo, isto é, a tradução pode começar antes da transcrição estar completa (isto não acontece nos eucariotas, pois os processos são separados fisicamente. Excecionalmente ocorrerá se existirem no núcleo ribossomas que controlem a qualidade do RNA). Assim, o único ponto possível de regulação é, quase exclusivamente, a nível da transcrição. TRANSCRIÇÃO DIFERENCIAL Relaciona-se com a taxa de transcrição que ocorre. Se for transcrito muito RNA temos muita proteína, enquanto se for transcrito pouco RNA temos pouca ou mesmo nenhuma proteína. OPERÕES – PROMOTORES PROCARIÓTICOS Contrariamente aos eucariotas, nos procariotas o promotor é reconhecido pelo fator sigma, da RNA polimerase, que se liga a ele e desnatura a dupla hélice de DNA. A polimerase depois liga-se à cadeia simples e inicia a síntese de RNA, após libertação do fator sigma. Fazendo o alinhamento de várias sequências de genes da E. coli, podemos confirmar que as zonas em que há maior nº de nucleótidos em comum são as regiões -35 (TTGACAT) e -10 (TATAAT). Essas regiões são então chamadas de regiões de consenso. NOTA: Podemos ver na imagem que a sequência codificante do gene é a parte a azul (que só começará com o codão ATG), mas a transcrição inicia-se antes (em +1), numa região que permite a ligação ao mRNA (região 5’-UTR), que embora depois não seja traduzida, é bastante importante para a ligação aos ribossomas. Sequência de consenso: sequência ideal no gene para interação com proteínas de regulação (sejam elas fatores de transcrição ou fatores sigma). Engenharia Genética F2 26 Os genes dos procariotas são maioritariamente policistrónicos (podem atuar vários ribossomas simultaneamente e do mesmo gene podem surgir diferentes proteínas) e a sua regulação é feita de forma conjugada por operões, sendo um processo mais simples e apenas a nível da transcrição e da tradução. Operões (cluster): controlam a expressão de vários genes (que não façam sentido ser expressos uns sem os outros) – genes policistrónicos – de modo a que esta seja apenas “ativada” quando o produto do gene é necessário à célula. São constituídos por um gene promotor, um gene operador e os genes estruturais. A estes liga-se um 4º gene, o gene regulador, que não faz parte da constituição do operão, mas funciona em parceria com. O gene promotor ou promotor próximal é a região (a curta distância do extremo 5’) onde a enzima RNA polimerase, responsável pela transcrição dos genes estruturais e fatores de transcrição, se liga. O gene operador é o que controla o acesso da RNA polimerase aos genes estruturais, regulando a sua transcrição, sendo o local onde se liga a proteína reguladora. Os genes estruturais são onde se encontra codificada a informação genética necessária para a formação de certas proteínas. O gene regulador vai ser constantemente transcrito e traduzido, produzindo continuamente pequenas quantidades de uma enzima proteica, o repressor. Esta enzima pode ser codificada na forma ativa (na qual se vai ligar ao gene operador, impedindo a passagem da RNA polimerase proveniente do gene promotor e, consequentemente, impedindo a transcrição dos genes estruturais) ou na fase inativa (na qual não se liga ao gene promotor, permitindo, assim, a passagem da RNA polimerase e a transcrição dos genes estruturais). A cada operão está associado um gene regulador que só produz repressores numa das formas, ativa ou inativa, nunca alternando entre elas. Engenharia Genética F2 27 Promotor lac (lactose) É dos promotores mais usados porque é facilmente regulado. A lactose é um açúcar usado pelas bactérias para obter energia. A E. coli necessita de sintetizar três enzimas (proteínas) que ajudem no processamento da lactose, para que esta possa atravessar a membrana citoplasmática. Na ausência de lactose, o gene regulador produz um repressor na forma ativa, que se liga ao gene operador, impedindo a passagem da RNA polimerase e consequentemente a transcrição dos genes estruturais. Na presença de lactose, esta liga-se ao repressor originando uma alteração conformacional que o torna inativo, o que vai levar à sua desconexão do gene operador, permitindo deste modo a passagem da RNA polimerase e a transcrição dos genes estruturais, após a tradução dos quais serão produzidas as enzimas necessárias ao metabolismo da lactose, ou seja, para a sua degradação em glucose. Quando é adicionada ao meio, a lactose ativa a expressão dos genes – regulação positiva. A ligação da RNA polimerase ao promotor lac é fraca e requer frequentemente ativação pela CAP (Catabolite Activator Protein). Quando a concentração de lactose começa a baixar drasticamente, devido à ação catalítica das enzimas (que a metabolizam em glucose e ATP), a lactose desliga-se do repressor, que, ao voltar à forma ativa,liga-se novamente ao operador, bloqueando a transcrição do operão, garantindo uma poupança de recursos que não são necessários na ausência de lactose. Como alternativa pode-se usar IPTG que funciona, analogamente à lactose, como inibidor da proteína reguladora. Contudo, este composto não é metabolizável e por isso não gera energia, e a expressão dos genes nunca cessa. É uma via catabólica em que há produção de energia, e os genes catabólicos são sempre regulados pelos níveis energéticos, ou seja, pelos níveis de concentração de ATP e AMP cíclico (que são inversamente proporcionais). Assim, como o aumento da glucose leva ao aumento de ATP (e diminuição de cAMP), na presença de muita glucose a expressão dos genes é inibida porque há demasiada energia. NOTA: A célula precisa de um nível de energia elevado para expressar o recombinante, mas se for muito elevado não o vai expressar porque não precisa. Por outro lado, se os níveis forem muito baixos a célula definha. É por isto preciso encontrar um nível intermediário adequado às condições experimentais em que trabalhamos. A lactose funciona como um indutor, pois a sua presença ativa o operão. É também por isso que se dá o nome de operão/promotor indutível. Engenharia Genética F2 28 Se houver tanto lactose como glucose no meio a bactéria não precisa degradar a lactose e por isso a expressão dos genes não é ativada – há portanto um duplo controlo do operão lac: Quando não há nem glucose nem lactose no meio, a CAP liga-se mas também o repressor se liga, pelo que continua sem haver expressão dos genes estruturais. Quando há glucose e não há lactose no meio, o operão está inativo porque há ligação do repressor ao gene operador e ainda porque a CAP não se liga. Quando há glucose e lactose no meio, a CAP não se liga e por isso não há expressão. Quando não há glucose mas há lactose, há ligação da CAP e da RNA polimerase porque o repressor é inativado pela lactose. Concluindo: este promotor é induzido por lactose e IPTG e amplificado pelo cAMP (e depleção de glucose). Promotor trp (triptofano) Os genes estruturais deste operão, quando transcritos e traduzidos, originam enzimas necessárias à produção do aminoácido triptofano, sendo que o gene regulador deste operão, contrariamente ao que acontecia no operão lac, codifica um repressor na forma inativa. Engenharia Genética F2 29 Desta forma, quando as concentrações intracelulares deste aminoácido são baixas, o repressor inativo, não podendo ligar-se ao gene operador, vai deixar o operão ativo, permitindo a passagem da RNA polimerase até aos genes estruturais e a produção das enzimas, levando, assim, ao aumento da concentração de triptofano. Quando a concentração deste aminoácido é elevada, algumas moléculas deste aminoácido ligam-se ao repressor, modificando a sua conformação e tornando- o ativo, pelo que ele se liga ao operador, bloqueando a transcrição dos genes estruturais do operão. Se houver cerca de 50% de triptofano no meio intracelular há uma atenuação da expressão dos genes, sendo que vai haver alguns fragmentos de RNA muito pequenos e algumas proteínas que não são expressas. Ou seja, em alguns casos a transcrição termina antes de atingir a sequência em hairpin. Como quando está no meio, o triptofano inibe a expressão dos genes estruturais deste operão diz-se que faz uma regulação negativa. Concluindo: este promotor é induzido por depleção de triptofano e presença de ácido 3- indolacrílico, e reprimido pela presença de triptofano. Promotores tac e trc Enquanto os promotores lac e trp eram promotores naturais nativos que existem na E. coli, os promotores tac e trc são híbridos artificiais feitos em laboratório que não existem na bactéria selvagem. Estes vão permitir uma produção 3x superior ao operão trp e 10x superior ao lac. Estes vão ter uma região TATAbox (-10) igual à do operão da lactose e uma região CGbox (-35) igual à do operão triptofano. Isto vai permitir-lhes ter uma regulação fácil e produzir em grandes quantidades como o operão lac e, por outro lado, a capacidade “babosa” do operão trp que é constitutivo, ou seja, está constantemente a produzir (a menos que seja reprimido). OPERÕES – PROMOTORES PROCARIÓTICOS FÁGICOS Os vírus são parasitas obrigatórios de outras células, sendo os específicos das bactérias chamados de bacteriófagos. Os promotores destes bacteriófagos acabam por ser mais fortes que os das próprias bactérias, e por isso é que estas são infetadas. A via anabólica consome energia, sendo uma via que está sempre ativa em contínua síntese, e que só para quando não é necessária devido à presença excessiva do seu próprio produto. A expressão destes genes é constitutiva, ou seja, contante, sendo que o triptofano atua como co-repressor. Este operão/promotor diz-se então reprimível. Engenharia Genética F2 30 Promotor pL: promotor de bacteriófago λ é regulado pela proteína codificada pelo gene CI, a qual é termosensível: a 30oC está ativa e liga-se ao promotor impedindo a transcrição dos genes, enquanto a 42oC desnatura e fica inativa, havendo expressão do promotor. A expressão dos genes estruturais deste promotor leva à formação de viriões. O que se faz é clonar em laboratório esta proteína juntamente com o promotor pL no qual está inserida a nossa proteína de interesse, em E. coli. Assim, quando se cultiva a bactéria a 30oC nada acontece e a bactéria cresce e a biomassa aumenta. A determinada altura, quando queremos que a bactéria comece a sintetizar o nosso produto, aumentamos a temperatura para 42oC. Promotor pT7: o promotor de bacteriófago T7 é apenas reconhecido pela polimerase fágica T7 RNA polimerase, e portanto nenhuma polimerase bacteriana o consegue reconhecer. Neste sentido, usando este promotor numa bactéria (por ser mais forte) tem-se de clonar também nela um gene codificante da polimerase fágica, o qual não precisa de ser muito tempo expresso porque a polimerase é extremamente ativa por apenas reconhecer um promotor. Desta forma, usa-se o gene lacI que vai codificar uma proteína que ao ligar-se ao promotor do gene codificante na polimerase fágica permite a sua expressão, e esta polimerase, por sua vez, vai induzir o promotor pT7 e permitir a expressão do nosso gene de interesse. Além da proteína lac também se pode usar IPTG para induzir a expressão da polimerase fágica. Este promotor pT7 é extremamente potente e permite desencadear um processo catastrófico de expressão, aumentando a quantidade de produto de interesse obtido. INDUÇÃO DA EXPRESSÃO/ AUMENTO DA PRODUÇÃO Repressores e promotores: quando a molécula repressora é muito forte é muito complicado induzir a expressão dos genes. Para solucionar o problema existem várias estratégias que podem ser aplicadas como reduzir o nº de cópias do gene codificante da molécula repressora no plasmídeo (ou cromossoma) ou aumentar o nº de cópias do gene de interesse. Em repressores pouco eficazes, o promotor é leaky, ou seja, está constantemente a exprimir os genes estruturais – é constitutivo. Se se quiser reverter a situação pode-se aplicar estratégias contrárias às indicadas para o repressor forte. Temperatura: se aumentarmos a temperatura as proteínas desnaturam e o promotor fica livre para poder produzir mais. Para aumentar a temperatura contudo precisa-se de uma fonte de calor com temperatura extremamente elevada, caso contrário o processo demora muito tempo. Além disso, o consumo energético é muito elevado e são necessários fermentadores especiais. É por isso necessário avaliar bem se o processo é economicamente rentável antes de o aplicar. Detergentes e promotores fágicos: a lactose é um indutor da expressão barato que, no entanto, não pode ser utilizada porque é metabolizada e satura o AMP cíclico. A alternativaé usar IPTG para exprimir os promotores da lactose que, no entanto, são “fracos” e acaba-se por micae Realce micae Realce Engenharia Genética F2 31 gastar muito dinheiro para exprimir pouco. Por este motivo é que se recorrem a promotores fágicos para controlar a expressão dos genes: um exemplo é a regulação em cadeia (trp+pL). (trp+pL): coloca-se o gene codificante da proteína CI acoplado a um promotor do triptofano e o nosso gene de interesse acoplado a um promotor pL, apenas ativado na ausência da proteína CI. À medida que a bactéria cresce vão surgindo cada vez mais promotores pL e por isso são necessárias mais proteínas CI para os inibir, basta então fazer crescer a bactéria num meio pobre em triptofano e assim o promotor trp ativo vai exprimir a proteína CI que por sua vez inibe o promotor pL e consequentemente a transcrição do nosso gene de interesse. Para induzir a expressão basta colocar a bactéria num meio rico em triptofano, o qual vai inibir o promotor trp e não se expressão a proteína CI. Desta forma, o promotor pL fica livre e o nosso gene de interesse é expresso, sem ser preciso um processo dispendioso de aumento de temperatura. Usando esta estratégia consegue-se 20% mais conteúdo proteico de interesse, o que é muito significativo. Múltiplas cópias do gene: quanto maior quantidade de genes, maior quantidade de RNA e maior quantidade de proteína sintetizada. Isto pode conseguir-se com múltiplas cópias do gene em tandem no plasmídeo e/ou com múltiplos plasmídeo em cada célula. Contudo, a relação nem sempre é de proporcionalidade direta e os resultados podem não ser os esperados, isto porque uma maior quantidade de DNA a expressar pode saturar energeticamente a célula e acabar mesmo por ser tóxico. Outra desvantagem é ainda a grande quantidade de cópias favorecer processos de recombinação genética entre elas e o aparecimento de mutações, o que diminui a estabilidade da proteína final, podendo mesmo obter-se um produto diferente do desejado. Aumentar a eficiência na tradução: existem duas maneiras fundamentais de aumentar a eficiência do processo de tradução para aumentar a produção da nossa proteína de interesse: A distância entre as regiões RBS (Shine-Dalgarno) e AUG (codão de iniciação) determina a ocorrência ou ausência de tradução, nomeadamente se estiverem muito afastadas o ribossoma desliga-se da cadeia de mRNA, e se estiverem muito próximas ele é capaz de não reconhecer o codão de iniciação, não ocorrendo portanto tradução. Outro aspeto que influencia é a estrutura secundária do mRNA, a qual determina a afinidade para com o ribossoma. Os codões do mRNA devem ser adaptados ao hospedeiro em causa, substituindo-os por aqueles que surgem em maior frequência no mesmo, ou seja, aqueles para os quais o hospedeiro sintetiza tRNAs complementares. Também se pode induzir a célula a sintetizar t-RNAs em falta em vez de alterar-se os codões. As adaptações nos codões são feitas por mutagénese dirigida ou por PCR. Na primeira muda-se apenas um nucleótido fazendo-se um primer de oligonucleótidos abrangente à cadeia de mRNA na região onde se quer inserir a mutação, mas o local central onde se encontra o nucleótido mutado não emparelha. A nova cadeia é depois sintetizada por complementaridade com a restante cadeia de mRNA. Desta forma vamos ficar com uma linhagem de plasmídeos mutada e outra não. A primeira permite obter uma grande quantidade de plasmídeos mutantes. Engenharia Genética F2 32 Minimizar precipitação: é muito complicado recuperar uma proteína depois desta precipitar. Para minimizar a precipitação podem usar-se proteínas integrais da membrana (como é o caso da tioredoxina). Folding e secreção apropriados: o folding da proteína pode não ser adequado ao hospedeiro, o que dificulta a sua secreção. Para melhorar o folding pode recorrer-se a proteínas DsbC (dissulfide bond-forming protein), que estabelecem pontes dissulfídricas entre cisteínas, garantindo que a proteína adquire uma estrutura correta. Consumos de oxigénio: clonando um gene, por exemplo o da hemoglobina bacteriana, juntamente com o nosso recombinante, consegue-se aumentar a capacidade do hospedeiro em captar oxigénio necessário às suas funções metabólicas, de modo a fazer face à expressão do nosso recombinante (processo que requer funções metabólicas mais exigentes). AUMENTO DA ESTABILIDADE DO NOSSO PRODUTO A proteína que recombinamos é um produto extremamente instável por ser estranho ao organismo hospedeiro em que é clonada, fazendo este de tudo para a degradar e eliminar. Existem, contudo, algumas estratégias para aumentar a estabilidade do nosso produto e, consequentemente, a sua durabilidade. Proteína de fusão: fundir com a nossa proteína uma proteína própria do hospedeiro (por exemplo, a β-galactosidase), colocando entre os seus genes uma região (7 aminoácidos) – linker de fusão – que funciona, após a expressão, como péptido-sinal reconhecido pelas protéases, separando assim no final o produto de interesse da proteína do hospedeiro. Esta proteína “extra” vai impedir a formação de corpos de inclusão destinados à destruição da proteína de interesse estranha ao organismo. Coloca-se primeiro a proteína recombinante ou primeiro a do hospedeiro? Depende da situação, tendo de se testar qual o melhor método, pois ambas as proteínas têm de estar na mesma grelha de leitura. Este passo pode mesmo afetar a estabilidade final do produto. Linker de purificação: além dos linkers de fusão explicados anteriormente, existem também os linkers de purificação que permitem recolher a nossa proteína de interesse de entre os restantes constituintes da fermentação por cromatografia de afinidade, ficando o nosso produto ligado à coluna da fase estacionária. Um exemplo é a expressão de Il2, uma citoquina (proteína) humana do sistema imunitário que nunca havia sido possível produzir em bactérias até ser fundida com uma proteína bactéria por meio de um linker de fusão. Este linker vai ainda ser útil para remover a proteína de interesse do meio de cultura, sendo depois cortado por uma peptidase. Outra forma alternativa de conseguir produzir e purificar a nossa proteína de interesse numa bactéria é hibridá-la com uma proteína integral de membrana que prende a nossa proteína do lado exterior da membrana, não havendo assim uma secreção total dela. Isto leva a que a proteína fique relativamente concentrada no fermentador, não sendo necessário processar todo o meio de cultura, apenas a biomassa. Contudo, não é um método muito rentável. Engenharia Genética F2 33 Marcadores seletivos (antibióticos): o uso de antibióticos para manter o gene de interesse à escala industrial não é rentável pois além de constituir lixo que depois tem de ser processado, tendo um custo relativamente elevado associado, e de promover a ocorrência de bactérias resistentes a antibióticos, são reagentes que só por si são muito caros. Existem algumas soluções alternativas ao seu uso que são mais vantajosas: Limitar o número de gerações por ciclo de produção evitando assim a perda do plasmídeo potenciada por mutações decorrentes dos processos de replicação. Para isto é preciso estimar a taxa de perda do plasmídeo de interesse e determinar o ritmo de trabalho adequado para eu não haja perda do mesmo. É ainda extremamente importante manter uma vigilância da sequência codificante do gene ou proteína durante o processo para assegurar que ainda se tem. Integrar o plasmídeo no cromossoma do hospedeiro por recombinação homóloga. Contudo, a recombinação tem de ser feita com regiões que não sejam críticas ao crescimento do organismo em causa. Bancos de células: devido às mutações vistas atrás que ocorrem em todos os ciclos de replicação dos hospedeiros e que potenciam a perda do nosso gene de interesse, é aconselhávelrecorrer a bancos de células. Nestes bancos começa-se por obter uma célula com as características ideais e o nosso recombinante, tendo esta de ser exaustivamente bem caracterizada, a qual é depois clonada (faz-se uma cultura em massa) e no final congela-se o lote de células, todas com características iguais. Faz-se a cultura, congelam-se 1000 ampolas e usa-se uma para fazer outra cultura em massa. Desta 2ª congelam-se mais 1000 ampolas e usa-se uma para produção industrial. Desta forma, estes bancos vão assegurar 106 ciclos de produção controlados e idênticos, com conservação da estabilidade genética do recombinante. Isto assegura disponibilidade contínua de células em caso de acidentes ou situações indesejáveis durante o processo de produção industrial do nosso recombinante. Embora ocorra variabilidade genética que nunca se consegue erradicar, é sempre constante (o número de mutações não aumenta, é sempre constante). Aminoácido N-terminal: é o aminoácido N-terminal da proteína de interesse que é reconhecido e sinalizado com ubiquitina para futura destruição da proteína, podendo-se substitui-lo por um com maior tempo de semi-vida, o que aumenta a estabilidade da proteína e o seu tempo de vida. Célula hospedeira sem proteases: as protéases são o principal motivo de degradação da nossa proteína de interesse, e a sua ausência seria um fator impulsionador da estabilidade do produto. Engenharia Genética F2 34 PLASMÍDEO DE EXPRESSÃO PROCARIÓTICO Concluindo, um plasmídeo de expressão procariótico deve ter: Origem de replicação ou local de recombinação com cromossoma (para poder ser integrado no genoma do hospedeiro) para que se consiga manter o plasmídeo dentro do hospedeiro e se consiga passa-lo à descendência durante a replicação – no primeiro caso replica-se independentemente do hospedeiro e no 2º caso, dependentemente. MCS (Multi Cloning Site) nas três grelhas de leitura para que se consiga inserir o gene de interesse, bem como 3 codões de terminação para a transcrição. Promotor forte e regulável para conseguir controlar temporal e quantitativamente a expressão do nosso gene recombinante, uma vez que se trata de um objeto estranho ao hospedeiro. Quanto maior forte o promotor, maior a quantidade de produto formada, mas se for demasiado forte pode saturar energeticamente a célula, daí ser preciso regulação. Marcador de seleção que garantem vantagem evolutiva na presença do recombinante, para que este seja preservado pelo hospedeiro. Podem usar-se antibióticos (que como já vimos apenas são viáveis de usar a escala laboratorial devido aos elevados custos de compra e tratamento de efluentes) ou aminoácidos (recorrendo a técnicas de auxotrofia, como o triptofano). Sequência de péptido-sinal para secreção do produto. Sequência de péptido (tag) removível que tanto pode servir para aumentar a estabilidade do gene como para melhorar a purificação do produto por cromatografia de afinidade. Outros elementos relevantes de usar consoante o tipo de proteína recombinante que se deseja obter. MANIPULAÇÃO DE EUCARIOTAS Embora a transcrição seja um ponto importante na manipulação de organismos eucariotas, existem muitos mais pontos passiveis de ser regulados. A mais significativa nos eucariotas até é mesmo a nível do processamento pós-traducional que, contrariamente aos procariotas, nestes organismos é exaustivo devido à necessidade que a proteína produzida tem em migrar desde o local de síntese ao local de atuação. Folding: tal como nos procariotas, o folding da proteína pode não ser adequado ao hospedeiro, o que dificulta a sua secreção. Para melhorar o folding pode recorrer-se a proteínas DsbC (dissulfide bond-forming protein), que estabelecem pontes dissulfídricas entre cisteínas, garantindo que a proteína adquire uma estrutura correta. No entanto, estas pontes são mais consistentes nos eucariotas, originando estruturas moleculares diferentes. Processamento proteolítico: remoção de fragmentos internos da proteína – splicing. Glicosilação: nos procariotas não ocorre glicosilação, mas nos eucariotas sim, embora nem todas as proteínas sejam glicosiladas da mesma forma (depende do organismo em causa, o processo é diferente caso se trate de leveduras, insetos ou mamíferos). Apenas Engenharia Genética F2 35 as proteínas de membrana ou secreção que são sintetizadas no retículo endoplasmático sofrem glicosilação, pois são as únicas que passam pelo complexo de Golgi (local onde ocorre o processamento), dependendo o grau de glicosilação do tempo que a proteína demore a passar esse organelo, ou seja, o tempo que demore a ser secretada até à membrana ou para fora dela (embora a glicosilação seja maior nas leveduras). A glicosilação pode ser feita na extremidade O- (por trionina ou serina) ou na extremidade N- (por aspargina). Modificação de aminoácidos: por fosforilação, acetilação, etc.. A transformação consiste na introdução de DNA estranho em bactérias ou leveduras por alteração das propriedades de crescimento em células animais. Este ultimo caso pode, contudo, levar ao desenvolvimento de tumores, o que limita o seu uso em humanos. À transformação está também associada uma alteração do genótipo do hospedeiro com consequente alteração no fenótipo. Transfeção: introdução de DNA em células animais. Além do DNA de interesse pode haver alteração de outros genes do hospedeiro (alteradas por vírus). PLASMÍDEO DE EXPRESSÃO EUCARIÓTICO Um plasmídeo de expressão procariótico deve ter: Origem de replicação (2) para E. coli (porque toda a manipulação do DNA tem de ser feita em E. coli) e para as células eucariotas em questão. Gene de resistência a antibiótico (não se usa em células eucariotas, mas pode ser relevante aquando da manipulação em E. coli) ou que permita seleção metabólica para os eucariotas (ESM – marcador de auxotrofia). A este último tem ainda de estar associado o seu promotor e terminador para poder ser expresso. MCS ladeado pelo promotor eucariótico e um terminador (essencial porque se a transcrição não terminar pode tornar-se tóxica para o organismo). Existem 3 hospedeiros eucariotas mais significativos na recombinação genética (por ordem de relevância, usando-se uma apenas quando a anterior não satisfaz os requisitos): saccharomyces cerevisiae (levedura alimentar), baculovírus (vírus de insetos) e células de mamíferos. Vamos então ver como podemos manipular cada um deles. SACCHAROMYCES CEREVISIAE Levedura (sistema) unicelular alimentar, genética e fisiologicamente bem conhecido. É um microrganismo GRAS (General Recognized As Safe). Tem um crescimento rápido (embora não tão bom como E. coli) e é pouco exigente quanto ao meio. Tem promotores fortes e regulados disponíveis e um processamento pós-tradução eucariótico. É uma célula pouco secretora, o que constitui uma desvantagem porque geralmente quer-se secretar o nosso produto. Mutantes auxotróficos. Engenharia Genética F2 36 Existem vários promotores para S. cerevisiae disponíveis, tanto constitutivos como indutáveis, para utilizar, sendo regulados por diversas maneiras consoante as suas condições de expressão. Atualmente já se fazem vários recombinantes nestas leveduras: para vacinas (hepatite B), diagnóstico clínico (hepatite C e HIV) e terapia humana (fatores de crescimento e insulina). Existem ainda vários tipos de plasmídeos e cromossomas que podem ser usados neste organismo. YEp (episomal): é um episoma que usa uma estratégia idêntica à expressão de plasmídeos em procariotas, mas em procariotas. Podem existir na forma livres ou integrados no genoma celular. Divide-se independentemente e não é preciso preocupar com os centrómeros. Têm grande instabilidade principalmente a longo prazo porque não têm estruturapara reconhecer os centrómeros e vamos ter várias leveduras sem ele – perda de expressão do episoma. Pode haver ainda reversão do fenótipo auxotrófico. A levedura preserva só o marcador de seleção, tornando-se mais vantajosa do que as recombinantes. YIp (plasmídeo de integração): estes plasmídeos não possuem origem de replicação mas sim regiões homólogas ao cromossoma da levedura que permitem, por recombinação genética, a integração do gene de interesse e do marcador de seleção no genoma do hospedeiro. Esta estratégia é a escolha nº 1 quando se trata de trabalhar em leveduras (embora também seja muito usado em células de mamífero). YAC: ver apontamentos em “tipos de vetores de clonagem”. Trata-se de um cromossoma artificial de levedura que são usados quando o fragmento de DNA é muito grande. PICHIA PASTORIS Nem todas as proteínas podem ser produzidas na S. cerevisiae devido às diferenças existentes a nível das modificações pós-traducionais. Como alternativa, ocorreu a P. pastoris. Levedura unicelular. Tem elevada densidade no crescimento, uma vez que não produz etanol (fator limitante do crescimento como produto de excreção). Não permite glicosilação das proteínas Microrganismos metilotrófico, ou seja, consegue crescer em meios de cultura contendo apenas metanol como única fonte de carbono e energia. O metanol é tóxico para o Homem. Engenharia Genética F2 37 Esta levedura possui um gene AOX (álcool oxidase) que, não sendo expresso constitutivamente, é induzido pelo metanol, sendo alvo para recombinação genética – coloca-se o nosso gene de interesse (GOI na figura) no meio do gene AOX. O plasmídeo desta levedura tem ainda: Gene de seleção auxotrófica de histidina (HIS) Gene de seleção por antibiótico (ampicilina) BACULOVÍRUS Crescem em suspensão. Vírus infecioso de células de inseto de várias espécies. É o sistema biológico com produtividade mais elevada associada, uma vez que a sua natureza consiste em infetar as células e produzir enormes quantidades de viriões (extremamente resistentes por terem uma cápsula proteica), e por isso vai também produzir grandes quantidades do nosso recombinante. O ciclo viral infecioso dos baculovírus pode ser feito por gemulação outra vez de corpos de inclusão intracelulares de poliedrina. Esta proteína atua como um “cimento” que protege os viriões formados, tendo um promotor de expressão associado extremamente forte. Contudo, a poliedrina é inútil em laboratório porque podemos controlar os vírus, pelo que o seu gene codificante pode ser substituído pelo nosso gene de interesse, ficando ele associado ao promotor forte da poliedrina. Para se conseguir construir um baculovírus recombinante é necessário recorrer a vetores de transferência que possuem o nosso gene de interesse e regiões de homologia com o DNA viral. Ao emparelharem é possível, por recombinação, exprimir o recombinante no vírus em detrimento do gene da poliedrina As únicas desvantagens desta técnica é o facto de não haver meio de seleção dos recombinantes, tendo-se de observar ao microscópio e selecionar as células que não apresentem formação de corpos de inclusão, o que significa que o gene foi recombinado. CÉLULAS DE MAMÍFERO Usadas quando se quer produzir proteínas humanas, uma vez que apresentam modificações pós-traducionais completas e adequadas ao ser humano. São mais exigentes a nível dos meios de culturas. Crescem em aderência, e por isso tem associados desperdícios de meio de cultura e um crescimento mais lento. Podem usar-se vários marcadores de seleção, não sendo isso uma limitação. Engenharia Genética F2 38 ELEMENTOS DE CONTROLO DA TRADUÇÃO Consoante o tipo de célula em questão, diferente tem de ser a expressão da proteína aquando da tradução, sendo preciso em eucariotas uma seleção rigorosa que não existe noutros hospedeiros, dos elementos de controlo da tradução, tendo estas regiões de ser controladas. Sequências UTR nas extremidades 3’ e 5’ que não são traduzidas têm de ter dimensão adequada para que o ribossoma faça um reconhecimento correto do gene. Sequência de Kozak análoga à de Shine-Dalgarno nos procariotas, que permite o reconhecimento do codão de iniciação AUG pelo ribossoma. Sequência de sinal para secreção da proteína Tag (T) para purificação da proteína Sequência de protéase (S) necessária para remover o tag após recuperação da proteína. Codão stop (SC) para parar a tradução, senão o processo torna-se tóxico para a célula. Além destas tem ainda de existir local de ligação das poli-A polimerase para que ocorra pooliadenilação. PROTEÍNAS MULTIMÉRICAS Este conceito é aplicado tanto a células procariotas como eucariotas. É possível produzir duas proteínas diferentes na mesma célula usando: Dois vetores na mesma célula: usar dois plasmídeos, um com cada gene codificante para uma proteína, tendo estes de ter marcadores de seleção diferentes. O problema é não haver produção equivalente das duas proteínas devido aos processos de recombinação que podem levar à perda de um dos vetores por recombinação ou a mutações que alterem a sequência. Isto origina diferentes números de cópias de cada plasmídeo e leva à sobreprodução de uma das proteínas em detrimento da outra, e por isso não se usa. Dois genes num vetor: o DNA considera-se bi-cistrónico e os genes são independentes, ou seja, têm dois promotores diferentes. O facto de haver dois promotores pode levar a sobreposições entre eles, não havendo garantias de expressão a nível de ambos os genes. Embora seja um método viável, também não é muito fiável. Dois genes num vetor separados por IRES (Internal ribosomal entry site): consiste basicamente em colocar uma sequência reconhecida pelo ribossoma (de origem viral) entre os genes, permitindo que dois ribossomas atuem e que haja uma expressão equilibrada de ambas as proteínas. É o método mais usado industrialmente. Engenharia Genética F2 39 INTEGRAÇÃO ESPECÍFICA/SELEÇÃO A integração específica do plasmídeo recombinante no cromossoma da célula através de regiões homólogas que recombinam. As células recombinantes podem depois ser selecionadas através de um método de seleção positiva usando neomicina – no qual se selecionam as células de interesse por resistência à neomicina concebida aos recombinantes pela inserção de um gene simultaneamente ao nosso gene de interesse – ou de seleção negativa usando ganciclovir – no qual selecionam-se as células não recombinadas. KNOCK-OUT Consiste na eliminação de genes da célula eucariota através de recombinações entre o DNA cromossomal e plasmídeos recombinantes. Os genes ficam assim “silenciados” e deixa de haver produção de uma determinada proteína por substituição da sua sequência no gene – é um método contrário à expressão do gene de interesse. Engenharia Genética F2 40 Engenharia Genética F2 41 III - APLICAÇÕES NO DIAGNÓSTICO CLÍNICO OS 5 FFS – APLICAÇÕES “Food”: alimentação humana e animal; engloba animais mutificados, alimentos transgénicos (GM), etc.. Os alimentos transgénicos, ao contrário do que se pensa, têm várias vantagens económicas, pois há menos desperdício alimentar devido à maior resistência do produto a ataques de pragas e herbicidas. O milho transgénico é o alimento mais relevante neste meio. Ele possui uma toxina de bactéria que permite o controlo de infestação por larvas. Todo o milho doce comercializado atualmente é transgénico, pois só assim pode haver tão grande resposta ao consumo. Outro exemplo é a soja (possui gene de resistência a herbicidas), a batata (relevante devido ao elevado consumo; o OGM é resistente a infestantes virais), mandioca (alimento rico em cianeto que pode ser tóxico para o ser humanoque, por ser muito usado em África, é importante a obtenção de um GM com menor teor em cianeto). A modificação de animais consiste em organismos maiores, com características de interesse mais acentuadas, etc.. Estes animais ainda não se encontram no mercado por não serem rentáveis, uma vez que o seu crescimento não é normal e alguns acabam mesmo por morrer a meio do processo por causa da inadaptação do seu corpo às circunstâncias a que são submetidos. “Fiber”: vestuário; engloba fibras não sintéticas (biológicas) como a seda, existindo uma intervenção sobre os animais/plantas para que estes produzam fibras mais vantajosas, a nível da qualidade e do custo monetário. O algodão transgénico possui um gene de resistência a infestação de larvas. Existem ovelhas transgénicas às quais são administradas genes adicionais de síntese da cisteína (aminoácido que devido à sua pouca disponibilidade limita o crescimento do pelo) levando a uma produção mais significativa de lã. Fibras de celulose são usadas ainda no vestuário. Este açúcar está presente na parede das células vegetais, sendo possível manipula-las. O ser humano não tem as enzimas necessárias para digerir a celulose (como a glucose no amido), mas os ruminantes sim porque têm bactérias celulásicas na flora estomacal. Essas mesmas bactérias podem ser usadas na degradação da celulose a nível industrial neste mercado, havendo fibras de celulose revestidas com fungos. “Fuel”: os combustíveis fósseis são limitados e um dia esgotarão. O uso destes combustíveis resulta na libertação de CO2, processo que também ocorre em vários seres vivos. Neste seguimento já há estudos que levam a pensar que as células, que conseguem fazer esse mesmo processo biológico, poderão ser uma alternativa aos combustíveis fósseis. Contudo, para a evolução deste mercado tem de haver uma maior carência de combustíveis devido a questões económicas e sociais, o que ainda não se verifica, levando, por enquanto, a uma estagnação nesta área. Etanol: gasolina + etanol (90:10). Metano: resíduos orgânicos do processo fermentativo em anaerobiose levado a cabo por microrganismos. Engenharia Genética F2 42 Glucose < Celulose: a celulose é degradada em glucose por celulases, a qual é depois transformada em etanol. Hidrogénio: as hidrogenases bacterianas retiram o hidrogénio da água, ou seja, a energia, a qual pode ser canalizada como “combustível”. “Feedstock”: aplicações ambientais e de biorremediação, no âmbito em que a biotecnologia pode fornecer métodos de biorremediação no tratamento da poluição que, atualmente, satura o meio ambiente. EnviropigTM: processo de eliminação do fósforo das fezes dos porcos por criação de animais transgénicos, permitindo fazer cultura de porcos em meio urbano sem incômodo para a população devido aos maus odores. Não está ainda a ser implementado por questões económicas. Bio-lixiviação e bio-oxigenação, bem como tratamento de efluentes são processos que atualmente são químicos mas que, com recurso à engenharia genética, podem ser tornados biológicos. No primeiro caso consistirá na extração não química de minérios, enquanto o segundo trata a degradação aeróbia e anaeróbia de águas residuais, gases e óleos. “Pharmaceuticals”: engloba terapias humanas, medicamentos/ vacinas e diagnósticos. É o mais relevante dos dias de hoje, sendo que 9/10 medicamentos mais vendidos são de origem biotecnológica. Existem atualmente mais de 150 medicamentos autorizados e em uso clínico e cerca de 500 em fases terminais de desenvolvimento. Medicamentos: os biológicos vão interferir com o sistema imunitário. Consistem em: Substâncias para controlo das respostas imunes (novidades absolutas); Drogas anti-virais e anti-tumurais (os tumores/ cancros consistem num crescimento incontrolável de células que o sistema imunitário não consegue restringir, sendo que os medicamentos vão mesmo atuar nele, ajudando-o). Só são viáveis enquanto a vida humana valer mais que o tratamento, pois são métodos muito dispendiosos (ex.: para a Hepatite C, 2 semanas de tratamento custam 70 mil euros). Novas vacinas (a vacina para a Hepatite B só é possível devido à engenharia genética). Meios de diagnósticos precoces, a nível da genética. Terapias genéticas: corrigem situações anómalas para que não nasçam crianças com deficiências (que “impedem” a evolução da espécie). Esta técnica vai contrariar a natureza humana e utilizar proteínas de substituição e “transgénese” de células somáticas, havendo ainda muita controvérsia ética. Clonagens xenogénicas: crescimento de órgãos e tecidos em animais para transplante humano, que elimina o problema de escassez de oferta por dadores humanos. Produtos individualizados: por exemplo, se alguém tiver um cancro extrai-se as células desse individuo que se “curam” por recombinação genética e voltam a ser administradas. São métodos que são únicos para o individuo doente em questão e não generalizados para uma certa doença. Este processo ainda não é viável atualmente. Engenharia Genética F2 43 APLICAÇÕES NO DIAGNÓSTICO CLÍNICO – DIAGNÓSTICO MOLECULAR TÉCNICAS IMUNOLÓGICAS Anticorpos Monoclonais São anticorpos homogéneos, com especificidade pré-definida e produzidos em larga escala, sendo obtidos a partir da imortalização de linfócitos B produtores, clonados e expandidos em linhas celulares contínuas ou, mais atualmente, por expressão de DNA recombinante em células eucarióticas. Um organismo produz anticorpos diferentes em tempos diferentes, sendo que o mesmo antigénio não provoca sempre a formação do mesmo anticorpo. Os anticorpos monoclonais, por sua vez, são anticorpos pré-definidos que têm sempre a mesma especificidade. Ou seja, são anticorpos produzidos por um único clone de um único linfócito B parental, o qual é clonado e imortalizado, produzindo sempre os mesmos anticorpos em resposta a um agente patogénico. Estes anticorpos são iguais entre si na estrutura, nas propriedades físico-químicas e biológicas, na especificidade e na afinidade, ligando por isso sempre o mesmo paratopo ao mesmo antigénio. A forma geral de fazer anticorpos monoclonais é feita apenas em ratinhos BalbC porque apenas há células do mieloma histocompatíveis com células destes animais: 1) Imunizam-se os ratinhos BalbC com os antigénios de interesse e recolhem-se as células do baço, 2 dias após a última injeção de antigénios, que é quando há um máximo de concentração de IgG (quanto mais imunizadas forem as células, melhores serão os anticorpos). 2) Inoculam-se as células do baço com células do mieloma (plasmócito tumoral), havendo fusão entre as duas com ajuda de PEG, gerando assim células hibridas que são mutadas – hibridomas. Este hibridoma possui as propriedades de crescimento das células imortais do mieloma e segrega o anticorpo específico produzido pelas células B do ratinho, permitindo assim o cultivo destas células indefinidamente e a contínua segregação de imunoglobina. É de notar que o anticorpo provém apenas das células do baço imunizadas. 3) Seleção metabólica dos hibridomas, que é necessária porque vamos ter: a. Células de mieloma fundidas umas com as outras. b. Células de mieloma não fundidas. c. Células de linfócitos B fundidas umas com as outras. d. Células de linfócitos B não fundidas. e. Células de linfócitos B fundidas com células de mieloma Só estas é que têm capacidade de crescer em ambos os meios. Para tal vai-se usar um meio de seleção HAT (hipoxantina, aminopterina e timidina) em que as células do mieloma não crescem de forma abrupta que elimine as células dos hibridomas, e que também não permita o crescimento de células dos linfócitos B não fundidas. » As células do mieloma expressam a enzima HPRT que catalisa a transformação de hipoxantina a purina, bem como as enzimasTK e DHFR. Estas enzimas possibilitam a via de síntese de purinas e pirimidinas essenciais à formação de ácidos nucleicos. A aminopterina do meio HAT bloqueia essas vias de síntese Engenharia Genética F2 44 por mutação das células do mieloma que leva a uma inibição das enzimas. Incapazes de fazer síntese de DNA, estas células acabam por morrer. » As células B acabam por morrer porque têm um tempo de vida limitado. » Os hibridomas, embora não tenham enzimas TK e sofram de bloqueio da DHFR, dividem-se normalmente por terem atividade da HPRT do linfócito B parental, podendo assim sintetizar DNA. Assim, ficamos apenas com as células dos hibridomas em cultura. 4) Seleção imunológica: embora no meio de cultura fiquemos apenas com hibridomas a crescer, estes possuem 4n cromossomas (por serem a junção do conteúdo de duas células diferentes). Contudo, durante a replicação o fuso acromático apenas tem possibilidade de se ligar a 2n cromossomas, havendo por isso cromossomas que não se ligam e não vão passar à descendência. Há assim a possibilidade de haver hibridomas que não possuam os anticorpos por perda dos genes que os codificam, o que implica fazer uma segunda seleção. Para isto faz-se uma placa ELISA em que se começa por forrar todos os poços com antigénios. Em seguida coloca-se em cada poço sobrenadante das culturas dos hibridomas. Os poços em que houver reação são os que possuem anticorpos e vão ser esses que vamos guardar por congelamento. 5) Clonagem: a única forma de garantir que não perdemos os anticorpos no futuro é clonar as culturas que sabemos que os possuem (fazendo- se cultura de modo a ter 1 célula em cada 3 poços). Temos sempre de testar a presença de anticorpos em ELISA e vamos assim continuamente selecionando e clonando, pois não temos forma de guardar as que produzem sempre anticorpos (não temos nenhum marcador), apenas temos meios de eliminar aquelas que não os produzem – controlo negativo. 6) Cultura das células: quando as células crescem segregam os cromossomas e ficamos com os anticorpos. Técnica de ELISA (Enzyme Linked ImmunoSorbant Assay) É um ensaio imunológico feito em microplaca (de 96 poços) que permite detetar e quantificar proteínas, péptidos, anticorpos e hormonas, sendo muito usado no diagnóstico de várias doenças que induzem a produção de imunoglobinas. Nesta técnica o antigénio é imobilizado numa superfície sólida, lava-se com tampão de lavagem, e depois é complexado com um primeiro anticorpo (o soro que queremos testar), lavando novamente a placa. Engenharia Genética F2 45 Posteriormente é colocado um tampão de bloqueio para preencher os espaços vazios entre os antigénios e evitar resultados falso positivos ou negativos, e a placa é novamente lavada. Em seguida adicionam-se os segundos anticorpos, aos quais está acoplado um enzima, e lava-se novamente a placa. Por último, adiciona-se o substrato, o qual vai reagir com o enzima e produzir produto mesurável, nomeadamente um produto colorido, com capacidade de se medir a absorvância – esta reação só ocorre se houver ligação antigénio-(1º)anticorpo. TÉCNICAS DE DNA RECOMBINANTE Hibridações moleculares A hibridação molecular mais significativa é a dos ácidos nucleicos. Quando fazemos uma eletroforese separamos os fragmentos de DNA. Imaginemos que queremos saber em qual dos fragmentos se encontra um determinado gene (vamos chamar-lhe “gene-alvo”) A hibridação de DNA permite identificar fragmentos de DNA que têm esse gene-alvo. Para isso usa-se uma sonda correspondente a uma cadeia simples com nucleótidos complementares ao gene-alvo, a qual se tem de marcar (radioactivamente ou recorrendo a outros métodos, como fluorescência) para se poder ver e seguir até esta identificar o gene-alvo por ligação a este por complementaridade de bases numa região. Para esta identificação ser possível é necessário o gene-alvo ter só uma cadeia de DNA (e não estar em cadeia dupla), o que é conseguido por desnaturação das moléculas de DNA com o aumento da temperatura. Como o aumento de T derreteria o gel usado na eletroforese, é necessário primeiro transferir o DNA para um membrana – a sonda não pode ser aplicada no gel. Faz-se então Southern ou Northern blotting. 1) Ligar DNA (ou RNA) alvo num suporte sólido (membrana de nitrocelulose ou plástico) 2) Adicionar DNA marcado (sonda radioativa, bioluminescente, etc..) – a marcação antes era feita com isótopos radioativos, mas atualmente é feita por bioluminescência. 3) Incubar em condições para hibridação para assegurar que a sonda se liga ao ácido nucleico por pontes de hidrogénio – manipula-se a temperatura e força iónica para alterar a ligação das moléculas, e desta forma a sonda pode ser usada várias vezes. 4) Lavar material não hibridado 5) Revelar sonda retida no suporte por autoradiografia, avaliação de cor, etc. – o tipo de revelação feito depende do tipo de marcação da sonda. NOTA: Se for feita pelo método normal demora algum tempo, mas atualmente já se consegue saber a resposta em 10-15 minutos. NAT (Nucleic Amplification Techniques) Engenharia Genética F2 46 DIAGNÓSTICO DE INFEÇÕES O diagnóstico de infeções é muito facilitado pelos métodos moleculares, havendo vários métodos: Método Vantagens Inconvenientes Exame microscópico Simples Deteção direta Discrimina organismos pela morfologia Lento, trabalhoso e repetitivo Baixa sensibilidade Não discrimina organismos semelhantes Perícia elevada Cultura in vitro ou inoculação animal Só deteta viáveis Avalia virulência e infecciosidade Lento e dispendioso Possível perda de viabilidade Uso de animais Existem microrganismos que não crescem nestas condições Deteção de anticorpos no soro Simples e rápido Automatizável Grande número de amostras Nem sempre específico Não discrimina infeção ativa de latente ou resolvida Hibridação de DNA e PCR Rápido, sensível e específico Deteta microrganismos diretamente e discrimina estirpes Independente da viabilidade biológica Automatizável Caro e dispendioso Não discrimina entre viáveis e não vivos Falsos positivos e falsos negativos. Estes falsos diagnósticos levam a tratamento que podem ser caros e/ou invasivos sem necessidade Por exemplo, um dos maiores problemas a nível nacional é a hepatite C que geralmente é transmitida em transfusões de sangue. Isto continua a ser um problema porque fica caro analisar o sangue a doar por PCR, e é uma doença que não é detetada pelos anticorpos. Estreptavidina/biotina: às sondas moleculares pode-se ligar covalentemente moléculas de biotina que têm capacidade de sequestrar e ligar-se a uma molécula de estreptavidina. Esta, por sua vez, consegue ligar a mais três biotinas, mas marcadas com a enzima fosfatase alcalina. Ao adicionar substrato, a enzima hidrolisa-o e há mudança de cor, que pode assim ser detetada. Esta técnica permite uma amplificação do sinal de hibridação, melhorando assim a deteção. Deteção de mutações pontuais conhecidas em enzimas (que deixam de funcionar) Semáforos moleculares (beacons): consistem em oligonucleótidos que hibridam com certos ácidos nucleicos por complementaridade de bases. Esta complementaridade é feita através dos seus 15 nucleótidos centrais. Na extremidade existem duas cadeias de 5 nucleótidos que são complementares entre si. Estes semáforos moleculares têm um fluoróforo na extremidade 5’, ao qual é ligado covalentemente um corante fluorescente (F); e um corante quencher na extremidade 3’, não fluorescente (Q). Engenharia Genética F2 47 Quando o oligonucleótido está em estrutura loop (não está hibridado) a extremidade do fluoróforo e do quencher estão emparelhadas, e os corantes estão próximos um do outro, o quencherabsorve a emissão fluorescente feita pelo fluoróforo. Ao hibridar com um ácido nucleico, as extremidades do oligonucleótidos separam-se e o quencher afasta- se do fluoróforo, havendo emissão fluorescente. Isto só acontece se a ligação de hibridação for mais forte do que o emparelhamento entre as extremidades, ou seja, se tiver mais bases complementares. Estes semáforos permitem ainda descobrir se os indivíduos homozigóticos ou heterozigóticos, tendo primeiro de se avaliar a descendência e a transmissão dos genes. O semáforo emite depois uma luz amarela se for homozigótico wild-type e vermelha se for homozigótico mutante. Desta forma, se o individuo for heterozigótico, hibridaram ambas as sondas, e portanto emite-se luz amarela e vermelha. Enzimas de restrição: conhecendo o mapa de restrição da enzima e aplicando-as, se os fragmentos obtidos forem diferentes dos esperados, então há possibilidade da enzima ter alguma mutação. PCR: usa-se um primer para o PCR que coincida com a região de potencial mutação. Se houver mutação e o primer não hibridar, não haverá amplificação. Em outros casos poderá haver hibridação quando existe mutação. Aptâmeros: Os aptâmeros são pequenas moléculas (15 a 60 bases) oligonucleotídicas (de ribo- ou desoxiribonucleótidos) de cadeia simples (ssDNA ou ssRNA) e com uma estrutura particular (secundária e terciária) que permite interagir com um alvo devido à elevada afinidade com a estrutura do mesmo. Por serem tão pequenos comparativamente com outras moléculas biológicas, têm uma melhor penetração nos tecidos. Têm um comportamento semelhante aos anticorpos. Ocorrem numa sequência de 15 a 17 bases por genoma humano. São produzidos artificialmente e modificados para serem mais estáveis, mas podem ocorrer naturalmente in vivo. Não são biotecnológicos, são produtos químicos. Os aptâmeros têm uma estrutura complementar ao seu alvo e funcionam como DNA ou RNA antisenso, hibridando com mRNA ou gene e manipulando especificamente a sua expressão. Enquanto os anticorpos específicos de antigénios são obtidos expondo animais a esse mesmo antigénio e extraindo e imortalizando o linfócito produtor de anticorpos, a aquisição de aptâmeros Engenharia Genética F2 48 não recorre a animais (eticamente positivo) nem tem de esperar que se desenvolva uma resposta imunitária, sendo por isso também vantajoso. Os aptâmeros obtêm-se então por exposição de uma molécula-alvo (antigénios) a RNAs que se ligam a eles, sendo assim possível seleciona-los e depois extrai-los e purifica-los. Aplicações Permite a descoberta de novas drogas e o controlo da sua disponibilização no organismo por interação com recetores. Permite diagnosticar algumas doenças Funciona como ferramenta terapêutica Usado em bioimagiologia É uma alternativa a reagentes analíticos Detetor de perigo biológico Relevante na área da inspeção alimentar Permite controlar SELEX (Systematic Evolution of Ligands by Exponential Enrichment) Método químico que recorre a bibliotecas de ácidos nucleicos (neste caso de RNA) para selecionar aptâmeros e baseia-se em ciclos consecutivos de seleção e amplificação. A biblioteca consistirá num conjunto de sequências nucleotídicas aleatórias que vão corresponder aos vários aptâmeros possíveis de serem usados. Começa-se então por incubar a biblioteca de aptâmeros com os vários alvos que, neste caso, se tratam de antigénios e selecionar os aptâmeros que melhor reagem/ligam ao antigénio, os quais são amplificados e incubados novamente com os mesmos antigénios, voltando a ser selecionados. Faz-se então vários ciclos deste procedimento de modo a evoluir o RNA (aptâmeros) ligante, tornando-o assim cada vez mais específico para esse mesmo antigénio. Este é um processo análogo ao que acontece a nível do baço durante a resposta imunitária, mas reproduzido em tubo de ensaio. Os aptâmeros podem sofrer modificações pós-selex para melhor interagir com o alvo e para ganhar resistência a nucleases e assim serem mais estáveis. No final os aptâmeros selecionados são clonados e sequenciados, podendo depois ser usados em alternativa aos anticorpos monoclonais. Esta síntese química permite modificar o aptâmeros durante o ciclo de modo a refinar a resposta a antigénios, o que não é possível no processo de obtenção tradicional de anticorpos monoclonais. Após cada modificação é necessário testar novamente a especificidade para o antigénio, para garantir que não se perde a complementaridade que é o objetivo principal. Engenharia Genética F2 49 IV - PRODUÇÃO INDUSTRIAL O principal objetivo na produção industrial é otimizar o processo em termos de minimizar os custos associados e maximizar a produção. Neste sentido um maior rendimento é obtido quanto maior for a quantidade de produto formado em menor tempo possível e usando um volume de microrganismos o menor possível. Para tal, é preciso ter em conta e intervir a nível dos aspetos biológicos do sistema de expressão e das características funcionais dos equipamentos usados. Ótimas condições de expressão do produto final (proteína) seriam: Baixas exigências nutricionais, nomeadamente usando microrganismos autotróficos. Produtividade elevada conseguida com promotores de expressão fortes e estabilidade elevada. Esquema de purificação final do produto fácil, rápida e eficaz, com um produto final de um nível de pureza elevado. Temos ainda de ter consciência que a célula está a produzir uma proteína estranha que lhe traz gastos energético e, por vezes, nenhuma vantagem, o que leva a uma baixa estabilidade da proteína. Desta forma, a célula responde com a degradação ou precipitação intracelular da mesma, tendo-se de arranjar soluções para contornar o problema. Estratégias usadas são: Fundir a proteína de interesse com uma proteína estável própria do organismo. Promover a secreção e diluição da proteína no meio de cultura. Quanto às escalas de produção, existem 3: laboratorial (desde o eppendorf aos 5L), piloto (dos 20 aos 200L) e industrial (+200L). Não há, contudo, forma das experimentações laboratoriais serem completamente análogas às industriais, e por isso durante uma produção industrial é sempre necessário ir avaliando os ciclos de produção e adaptando as condições de produção em função dessa análise. Para uma expressão rentável da proteína em questão há que ter uma expressão elevada da mesma, de forma regulada e recorrendo a promotores moduláveis. PRODUÇÃO DE PROTEÍNAS RECOMBINANTES Existem 5 etapas principais na produção de proteínas recombinantes em duas fases: upstream – fermentação, recolha e concentração de células – e downstream – precipitação/centrifugação do produto e purificação cromatográfica. FERMENTAÇÃO – CURVA DE CRESCIMENTO DE UM MICRORGANISMO (em batch) Engenharia Genética F2 50 Fase lag ou fase de latência Não começa na origem porque não há geração espontânea, ou seja, no início temos sempre um nº mínimo de células na cultura. Primeira fase antes de se iniciar a divisão celular, não havendo variação do número de células, uma vez que é o período de adaptação das mesmas ao meio. Há, contudo, uma intensa atividade metabólica. Esta fase tem duração variável, pois depende principalmente de dois fatores, os quais podem ser manipulados a nosso gosto, de modo a alterarmos a duração desta fase. Esta fase pode mesmo não existir. Vai depender: Da natureza do meio de cultura (da adaptabilidade das células ao inóculo): se for diferente do meio onde as células se encontravam inicialmente, o tempo de adaptação vai ser maior (pois, nomeadamente, as células vão ter de sintetizar novas enzimas), o que também acontece se o inóculo for muito pequeno. Das condições do micróbio: da sua idade (a duração é menor se tanto as célulascomo os meios forem mais jovens, pois estas têm metabolismo mais ativo, o que diminui o tempo de atuação) e da temperatura (se a T do meio inicial for muito baixa, o tempo de adaptação é maior). Fase log ou fase exponencial Nesta fase as células já estão plenamente adaptadas ao meio, absorvendo os nutrientes e sintetizando os seus constituintes – crescimento equilibrado. Elas crescem e dividem-se à sua taxa máxima (e constante, uma vez que se duplicam em intervalos de tempo regulares), de acordo com: O seu potencial genético A natureza do meio As condições de crescimento A quantidade de produtos finais de metabolismo ainda é pequena – ocorre metabolismo primário (ex.: ácido acético). Esta fase ocorre até as condições do meio se comecem a deteriorar (acumulação de produtos do metabolismo, alterações no pH, etc.) e passem a ser inibitórias do crescimento. Fase estacionária Nesta fase o crescimento populacional estabiliza, e a curva fica horizontal (não há crescimento líquido celular). Isto porque o número total de células viáveis mantem-se constante enquanto a taxa de crescimento iguala a taxa de morte celular (é errado dizer que não há crescimento ou que as células estão todas mortas), ou então a taxa de divisão celular ocorrer muito lentamente. Isto acontece devido a: Limitação de nutrientes Limitação de oxigénio (no caso de serem aeróbios) Acumulação de produtos do metabolismo (efeito tóxico) Como o sistema é fechado não são removidos os produtos de excreção, que são nefastos à população. Se ter atingido um nível populacional crítico Causa menos comum. Acontece quando o crescimento metabólico é tal que as células ficam sem espaço para se reproduzir. Entre a fase estacionária e a fase exponencial existe um período de tempo correspondente a uma outra fase – fase de desaceleração, na qual o declive da curva fica menos acentuado. Se as condições forem ótimas/ideais, então a taxa de crescimento será mesmo a velocidade máxima que a genética lhes permite. Engenharia Genética F2 51 Tanto nesta fase intermédia como na fase estacionária, o metabolismo primário reduz e o metabolismo secundário fica ativo, como mecanismo de defesa celular, por produção de substâncias que permitam a sua sobrevivência (ex.: produção de antibióticos e enzimas). Estes metabolitos são explorados maioritariamente na indústria farmacêutica devido aos grandes custos de produção. Outras indústrias não os usam mesmo que tenham interesse, pois não têm retorno económico. NOTA: Se a escassez de nutrientes for muito grande entra-se em fase de senescência, e por isso a margem de concentração de nutrientes é muito apertada e por isso é mais complicado manter os microrganismos nesta fase, relativamente à fase log. Fase de morte Esta fase inicia-se quando não há mais nutrientes fornecidos ao sistema ou os metabolitos começam a matar as células. O número de células metabolicamente ativas (viáveis) decresce pois a maioria das células está em processo de morte. O nº de células pode manter-se constante porque há células que não lisam. As células que morrem podem servir de nutrientes para as que estão vivas. Fase logarítmica: Uma proporção constante de células morre em cada intervalo de tempo. Usualmente um declínio logaritmo não dura o resto do processo. Fase não logarítmica: Varia com as condições ambientais como o tipo de meio ou o microrganismo em causa. FERMENTAÇÃO – TIPOS DE CULTURAS Culturas contínuas: sistema aberto em que há constantemente fornecimento de nutrientes frescos e remoção do “meio velho”, sendo estes fluxos de entrada e saída iguais (igual velocidade). Permitem um elevado número de cópias dos genes, mas não é viável em produtos de recombinação genética pois tem associada uma grande instabilidade genética que pode levar à perda do gene de interesse por degeneração decorrente de mutações espontâneas ao longo dos ciclos de replicação – esta é a maior desvantagem das culturas contínuas; pode atingir níveis A taxa de morte pode alterar-se (reduzir-se) ao fim de algum tempo devido à existência de formas particularmente resistentes – Fase Senescente. Neste caso o declive da reta do gráfico diminui. Engenharia Genética F2 52 de toxicidade consequentes da acumulação de produtos da fermentação que inviabilizem a cultura por alterações do meio; sendo ainda difícil manter a esterilidade do processo. Contudo, estas culturas também apresentam algumas vantagens: Pode-se usar reatores mais pequenos Menores períodos de paragem Processamento posterior em menores volumes Condição fisiológica das células mais constante Culturas descontínuas (batch) e semi-contínuas (fed-batch): o mais rentável é usar culturas em fed-batch porque tem associada uma grande produtividade final. Contudo, em passos intermediários do processo de produção industrial podem-se usar culturas em batch que permitem obter uma grande quantidade de biomassa (embora em termos gerais, seja um processo mais desvantajoso que o fed-batch). Batch: sistema fechado em que não se acrescenta nem retira nada da cultura ao longo do processo (há, contudo, troca de gases) – há crescimento das células até esgotamento dos nutrientes. Fed-batch: começa-se com um meio de cultura descontínuo e, em determinado ponto do crescimento (à nossa escolha) é introduzido meio de cultura (mas não é removido nenhum caldo de fermentação), o que vai aumentar o volume (e por isso este é variável). Isto permite que as células continuem a crescer mesmo quando a densidade é muito grande, mantendo o crescimento exponencial durante um período extra de tempo, o que aumenta a produção de produto final – produz-se mais e durante mais tempo. Condições ideais da cultura Temperatura: maioritariamente a temperatura ótima de funcionamento das células (de mamíferos) é de 37oC. Fase de crescimento: a fase ideal para produzir o medicamento é fase de crescimento exponencial (fase log) tardia, sem que esteja mesmo em fase estacionária. Não se usam nunca, por isso, culturas contínuas mas sim em fed-batch, de modo a estabilizar a cultura momentos antes da fase estacionária (pode-se ainda fazer algumas culturas em batch, mas é menos usual). Agitação: tem de ser permanente suficiente para manter a cultura em suspensão e homogénea, uma vez que as células eucariotas são muito grandes e têm tendência a precipitar. No entanto, não pode ser muito violenta para que não haja rutura e morte celular, e ainda para evitar a formação de muita espuma (que para ser anulada têm de ser adicionados ao meio compostos específicos). Engenharia Genética F2 53 FERMENTAÇÃO – TIPOS DE REATORES Fermentadores com agitação mecânica (pás de rotação): não se usam para células eucariotas animais porque, como não têm parede celular, são muito frágeis, mas são ótimos para células procariotas. Contudo, por estas serem muito pequenas obrigam a uma velocidade de agitação muito elevada, o que leva à formação de espuma, prejudicial ao processo, não podendo acrescentar-se anti-espumantes porque prejudicam o crescimento celular (uma vez que levam à produção de álcool que é tóxico para a cultura em certos níveis de concentração). Fermentadores com coluna de bolhas: a agitação é feita por uma coluna de bolhas formada por um difusor de ar colocado no fundo do reator, o que diminui a tensão de corte e assim pode ser usado em microrganismos mais frágeis. Fermentadores com circulação por arejamento: consiste numa coluna de bolhas mas dividida por um anteparo, cuja colocação influencia se ocorre circulação do ar externa ou interna. Tal como a coluna de bolhas tem baixas tensões de corte, mas acaba por ser mais eficiente que esta. Entre as duas circulações possíveis, o loop externo é a mais eficiente.RECOLHA – INDUÇÃO DA EXPRESSÃO Pela temperatura: utilizam-se dois fermentadores com temperaturas diferentes, e ao passar as células do fermentador a menor temperatura para o de maior temperatura, as proteínas desnaturam e podem ser recolhidas, e o promotor fica livre para produzir mais. Não é um processo muito usado porque requer muito tempo e implica um consumo energético elevado e condições de fermentação especiais. Por detergentes: pode-se remover células com ajuda de detergentes que rompem as membranas e também desnaturam as proteínas. RECOLHA/CONCENTRAÇÃO DAS CÉLULAS A filtração simples é o método mais frequente em laboratório para recolher as células, mas não industrialmente. Geralmente faz-se uma centrifugação zonal associada a filtração tangencial, na qual o líquido vai passando pelo filtro e algum meio passa livre de células, enquanto as células são arrastadas e recolhidas por outra extremidade. Engenharia Genética F2 54 PURIFICAÇÃO DO PRODUTO A purificação do produto começa por submeter a mistura, após filtração para separação das células, a uma cascata de filtros com diferentes dimensões de poro. Após isto recorre-se a processos cromatográficos. Podem-se usar diversas cromatografias como as de afinidade, as de exclusão molecular e as de troca iónica (tanto catiónica como aniónica). As que são aplicadas dependem do tipo de produto que se pretende obter mas, geralmente, aplica-se primeiro uma cromatografia de afinidade seguida de duas cromatografias de troca iónica (uma de cada tipo) para limpar completamente as impurezas. Cromatografia de imuno-afinidade: é um processo altamente seletivo, permitindo obter proteínas com um grau de pureza 100x superior ao da amostra aplicada. Consiste na afinidade entre a matriz de suporte cromatográfico e os componentes a separar, sendo específica para um grupo de proteínas. A separação é feita em função do ligando: quando a mistura (meio onde se encontra a proteína de interesse, proteínas contaminantes e outros componentes) atravessa a coluna, apenas a proteína de interesse se liga à matriz. Isto acontece porque a resina usada para a fase estacionária forma ligações covalentes (fortes) com ligandos altamente específicos para essa proteína (que neste caso serão anticorpos específicos). Por sua vez, a ligação proteína- ligando é mais fraca para no final permitir uma rápida eluição da proteína, mas suficiente para a reter inicialmente. As restantes moléculas são eliminadas por lavagem da coluna com uma solução tampão, visto não se terem ligado fortemente à coluna. O enzima desejado é posteriormente retirado da coluna (com elevados níveis de pureza) através de alteração nas condições de eluição. Cromatografia de troca iónica: a separação é feita pela carga elétrica, sendo feita em coluna com recurso a uma matriz (resina) que é um polímero contendo grupos ligados carregados. Neste seguimento, a resina pode ser de permuta/troca catiónica ou de permuta/troca aniónica, sendo que haverá troca de catiões (carga positiva) ou aniões (carga negativa), respetivamente, com os grupos ionizáveis das proteínas, retendo-as. O pH influencia a carga da proteína. As proteínas têm um ponto isoelétrico (pI) que corresponde ao valor de pH para o qual apresentam carga elétrica igual a zero (equilíbrio entre cargas negativas e positivas dos seus grupos iónicos): na troca catiónica: pH < pI; na troca aniónica: pH > pI Quanto maior a diferença entre a carga da proteína e da matriz, mais depressa a proteína é eluída (porque não fica retida). A retenção dos diferentes compostos é feita pela carga elétrica dos aminoácidos das proteínas. As cromatografias são fáceis, baratas e permitem uma purificação significativa do produto. Engenharia Genética F2 55 ENRIQUECIMENTO/ACABAMENTO (POLISHING) DO PRODUTO FINAL No final, para refinar o produto final, pode-se fazer: Diálise e osmose reversa – permite mudança de tampão Filtração estéril (0,22µm/0,45µm) – para esterilização Ultrafiltração – para separação de moléculas (cut-off) Diafiltração – para mudança de tampão e concentração Nanofiltração (1nm) – para remoção de vírus Engenharia Genética F2 56 Engenharia Genética F2 57 V - ORGANISMOS GENETICAMENTE MODIFICADOS (OGM) PLANTAS TRANSGÉNICAS Pensando em plantas geneticamente modificadas pode-se pensar que é algo pouco natural, mas a verdade é que ocorre na natureza sem a intervenção humana. Um exemplo é o de uma planta que sofre uma mutação chamada Crown Gall, na qual parte do plasmídeo duma bactéria que a infeta, integra o genoma da planta infetada. Isto vai resultar na formação de um “tumor”. * As células vegetais são totipotentes, ou seja, qualquer célula pode originar uma planta inteira (nomeadamente transgénica) por clonagem. A transgénese vegetal tem vista, principalmente, em obter plantas: Resistentes a pragas, por introdução de genes “inseticidas” que codificam toxinas mortais para determinados seres vivos que as ataquem. Resistentes a pesticidas e herbicidas, por modificação da planta para que esta os tolere, como introdução de genes que codifiquem enzimas que metabolizem os compostos aplicados. Isto permite que os pesticidas possam ser administrados mais cedo (no ciclo de vida da planta), e por isso em menor quantidade, sem serem prejudiciais para a planta, o que torna a administração de pesticidas mais barata. A empresa Syngenta Novartis é a mais relevante atualmente na área dos organismos transgénicos. Os primeiros produtos transgénicos a surgir o foram: o milho, os pimentos amarelos e vermelhos, as maçãs, as uvas e as melancias. O milho é o mais relevante organismos transgénico atualmente, seguido do algodão e do salmão (que para ser consumido ao ritmo atual, só pode ser transgénico). Qualquer produto transgénico é submetido a ensaios de campo extensivos onde se testará a resistência, valor nutricional, digestabilidade, potencial tóxico e alérgico, etc., para garantir que podem ser usado para consumo humano a nível mundial e que não são prejudiciais ao ambiente. MILHO BT11 Até hoje, a planta OGM mais relevante é o milho doce (Zea mays) ao qual foi adicionado dois genes das bactérias do campo Bacillus thuringiensis (BT) e Streptomyces viridochromogenes. O primeiro confere-lhe resistência a pragas por insetos pois codifica uma proteína que se liga ao tubo digestivo dos insetos quando eles atacam a planta, o que lhes é letal. Como o tubo digestivo dos mamíferos não tem recetor para esta proteína, não são afetados. O segundo gene codifica uma proteína capaz de degradar glifosatos de amónia usados como herbicidas, tornando assim a planta resistente. Isto permitiu uma redução significativa das perdas anuais das culturas de milho, e um aumento da disponibilidade como recurso alimentar. Contudo, a aprovação deste milho foi um longo processo, desde 1998 a 2004: Engenharia Genética F2 58 1) Submissão do dossier em 1988 2) Em 2002: ocorreram de uma série de comités científicos (primeiros a avaliar a situação) que apoiaram a produção deste milho, frisando que não havia evidência deste provocar efeitos adversos na saúde humana e no ambiente. 3) Em 2003: Standing Committee, composto por 50% cientistas e 50% políticos são os segundos a avaliar, e neste caso abstiveram-se de apoiar ou condenar a produção do milho. 4) Em Abril de 2004: avaliação feita pelos Ministros da Agricultura, que se abstiveram. 5) Em Maio de 2004: Decisão do Parlamento Europeu que recusou a transação do milho transgénico, por não terem interesse em negociar com os USA. 6) Em Maio de 2004: Comissão Europeia toma a decisão final, a favor da produção e comercialização do milho transgénico (provavelmente porque começa a desenvolver-se na Alemanha, e já não há risco de terde importar dos USA). Os alimentos transgénicos regressam assim ao mercado europeu, embora algumas corporações ainda fossem contra ou não fossem totalmente a favor. No entanto, ocorreram uma série de incidentes que tem atrasado o mercado destes produtos transgénicos, tais como: Triptofano recombinante: o triptofano é um aminoácido essencial para a síntese proteica, não sintetizado pelo nosso organismo. Era por isso comercializado triptofano de extração como suplemento alimentar, o qual foi depois substituído pelo recombinante. Notou-se pela primeira vez que o triptofano possuía um contaminante que aumentaria a possibilidade de doença, e o produto foi abolido por ligarem o contaminante ao recombinante. O que se veio a descobrir foi que o triptofano de extração também possuía o mesmo contaminante, mas este já não foi proibido no mercado pois era necessário como suplemento. Batata transgénica (1988): durante um ensaio em que ratos foram alimentados durante 110 dias apenas com batata transgénica notou-se uma supressão da resposta imunitária dos mesmos, noticia que correu os media e alarmou contra os produtos transgénicos. Testou-se depois o que aconteceria se os ratos fossem alimentados apenas com batatas não transgénicas e obtiveram-se consequências idênticas, mas estas já não foram divulgadas pelos media. Borboleta monarca (1999): um estudo de laboratório gera alerta dos pólens de plantas transgénicas elevarem a mortalidade desta borboleta adorada pelos americanos, entre outros insetos, o que gerou polémica. Contudo, um estudo científico de 2 anos veio comprovar que a quantidade de pólen usado durante o estudo de laboratório tinha sido muito maior do que libertada em terreno e que o período de polinização não coincidia com o período de alimentação das larvas das borboletas, concluindo assim que não havia perigo para os insetos. Estes dados foram ignorados pelos media. Milho StarLink (2000): teria um gene de resistência ao calor e era usado apenas para alimentação animal. Em 2000 foi detetada contaminação generalizada de alimentos humanos com StarLink e este foi retirado do mercado. Um estudo de 2001 não conseguiu, contudo, detetar marcadores de StarLink nos alimentos. Milho MON863 (2005-2007): surge em 2005 milho transgénico com toxina modificada contra um certo inseto, cuja toxicidade hépato-renal (para o fígado) é contestada em 2007. Esta contestação é feita com base nos estudos feitos para a aprovação, mas manipulando os dados estatísticos (diferente seleção dos dados a analisar e uso de testes estatísticos diferentes) que, não provando existir realmente um problema, também não comprovam que seja 100% seguro, e por isso demandou-se a retirada do mercado. Engenharia Genética F2 59 ANIMAIS TRANSGÉNICOS Ao contrário das plantas, não existem muitos animais transgénicos. Isto acontece por vários motivos, nomeadamente o facto das células vegetais serem totipotentes e as animais não (ou pelo menos achava-se que não, porque atualmente já há tecnologia nessa área). Desta forma, embora se possam ter animais transgénicos, a descendência resultante dum cruzamento poderá não ser transgénica, a menos que se modifiquem os genes de ambos os progenitores. Diz-se que ocorre transgénese quando há alteração de genes (e consequentemente de fenótipos) na linha germinal, por adição ou eliminação/inativação de genes (knockout), havendo alteração do animal e da sua descendência. O principal objetivo dos animais transgénicos é gerar novas características, com preservação das precedentes (das que o animal já possuía antes de ser modificado), nomeadamente para: Aumentar rendimento de leite Modificar características da lã Aumentar taxa de crescimento Aumentar frequência das posturas de ovos, etc.. Antigamente já se fazia uma “certa recombinação genética” ao promover cruzamentos específicos com seleção dos progenitores. Contudo, este método é muito demorado e exigente e perdem-se características anteriores, embora seja bem-sucedido. No laboratório, a recombinação genética faz-se seguindo 4 passos gerais: 1) Gene clonado é inserido no núcleo (modificado) de um ovo fertilizado. A inserção do novo gene no património das linhas germinais pode ser feito por: Vetores retrovirais em embriões Micro-injeção de DNA em pro-núcleos, após fertilização. É o mais eficiente. Recombinação in vitro de células estaminais e reintrodução do blastocisto Transferência de núcleos de células somáticas em ovo anucleados 2) Ovos fertilizados são inoculados em fêmeas recetivas 3) Seleção dos membros da descendência que possuem a nova característica 4) Estabelecimento das novas linhas genéticas com transgénico estável A transgénese animal promete um melhoramento animal específico e, atualmente, já fornece modelos animais para o estudo de doenças humanas (como o caso dos murganhos, ratinhos geneticamente modificados usados em laboratório), e produtos pharming (produtos farmacêuticos de valor acrescentado incorporados em produtos animais, como o leite e os ovos que por serem extracorporais não afetam o animal). Esta última questão não está mais aprofundada porque os produtores de medicamentos sofreriam economicamente com ela, embora já se produzam muitas proteínas para uso terapêutico em glândulas mamárias. Existem ainda algumas questões técnicas associadas à transgénese animal, vamos ter: Local de inserção do transgene, pois este pode interferir com os outros já existentes, induzindo a sua rutura, a formação de tumores ou a indução de provírus (no caso do uso de vetores virais). Número de cópias inseridas ainda não se domina, e isso pode ser um problema. Envolvente genética para a expressão NOTA: Para obter descendência recombinante tem-se de fazer cruzamento entre animais iguais, mas nunca podem ser da mesma família porque isso homogeneizaria os genes, ou seja, perde-se a zigotia e a diversidade, podendo mesmo perder-se a espécie. Expressão nos tecidos adequados, e se for expresso em tecidos inadequados pode ser um problema. Engenharia Genética F2 60 Engenharia Genética F2 61 VI - REGULAMENTAÇÃO Existem diversas campanhas de opinião sobre os produtos biotecnológicos que têm em conta questões éticas, legais, económicas (mais relevante na sociedade atual) e sociais, sendo os alvos o público que não é contra nem a favor deles (considerando os que são contra uma “causa perdida”). Para que um produto biotecnológico seja libertado no mercado existe uma série de guidelines que têm de cumprir, sendo feito estudos intensivos sobre o seu impacto a nível ambiental e da saúde pública, e ainda considerações específicas individuais para cada produto (nomeadamente económicas). A nível ambiental preocupa a transferência horizontal de genes entre espécies relacionadas e não relacionadas e o aumento da resistência/tolerância de possíveis alvos do produto. A nível da saúde pública, além de todos os ensaios feitos previamente e dos estudos animais que se fazem, há que garantir rotulagem do produto para que a sua escolha seja livre, e que observar os indivíduos que usem os produtos para vigilância e garantia da sua segurança. As patentes são concessões públicas conferidas pelo Estado que garantem ao seu titular exclusividade ao explorar comercialmente a criação de um produto, e em contrapartida este tem de ser disponibilizado ao público. Para ser patenteado: Não pode existir antes nem ter sido publicado nada sobre si há pelo menos 1 ano. Não pode ser apenas uma descoberta, mas sim algo “não óbvio” para conhecedores da área. Deve ser útil e aplicável (ou seja, facilmente reproduzido). Têm de ter descrição completa e serem implementadas por um conhecedor da área. Tem de ser feito pelo Homem As patentes em biotecnologia são muito rentáveis. Um exemplo é a eritropoietina,hormona que aumenta a quantidade de glóbulos vermelhos e consequentemente a capacidade respiratória, sendo usada no tratamento de doentes com insuficiência renal ou como esteroide, e no início rendeu 4mil milhões de dólares, embora atualmente já não tenha tanta relevância. Há algumas coisas que não são patenteáveis: Teorias científicas Métodos matemáticos Criações estéticas Tratamentos ou terapêuticos de humanos ou animais Produtos da natureza Segmentos de DNA que supostamente não deveriam ser patenteados já o são, ou seja, se quisermos usar um gene inteiro, cDNA ou sequências parciais, mesmo que sejam nossas, tem- se sempre de pedir autorização. Outras coisas que já estão patenteadas são: Processo para produzir biologicamente quimeras moleculares funcionais (1980-1997). OncoMouse (1988) – ratinho que nasce com um oncogene sob promotor viral nas células germinais e por isso irão durante a sua vida desenvolver tumores em diferentes tecidos, sendo úteis para testar componentes causadores ou preventivos de tumores. Contudo existem certas questões éticas (sentida à vida, rotura da integridade das espécies, tratamento inumano a animais) que se opõe à sua utilização. OGM/Transgénicos Engenharia Genética F2 62 TRANSGÉNESE E CLONAGEM HUMANA Nos séculos XX e XXI evoluiu-se bastante na área da clonagem, sendo que em 2000 até de tentou trazer um animal em extinção noutra espécie, embora isso seja um processo complicado pois se houve extinção era porque não tinha adaptações suficientes às condições ambientais. Os genes são sequências de material hereditário (DNA) responsáveis pela expressão das características fenotípicas, sendo “imortais”. Por sua vez, os indivíduos são um produto biológico resultante de um processo evolucionista, hospedeiro temporário dos genes que os expressa fisicamente. Ou seja, a função do individuo é propagar os genes! Pode-se assim dizer que a clonagem (qualquer uma), sendo um processo no qual se favorece a preservação dos genes e se facilita a propagação do material genético, é pro-natura, pois constitui o objetivo da existência de vida! CLONAGEM HUMANA Contudo, a clonagem humana ainda não é aprovada e é condenada ética, mora e legalmente. As suas hipotéticas aplicações seriam: Para fins terapêuticos: resolveria o problema da falta de doadores de órgãos. O uso de células do sangue do cordão umbilical são uma alternativa à clonagem (podendo fazer-se uma terapia autóloga – o processo envolve só o individuo a tratar – ou heteróloga – o tratamento envolve mais do que um individuo). Características particulares: ajudaria a eliminar as doenças hereditárias e permitiria escolher características particulares, como a cor dos olhos, num hipotético filho. Com isto correr-se-ia o risco de diminuir a diversidade genética e até extinção do Homem. Substituição de indivíduos: substituir entes queridos que morram ou prolongar a vida daqueles que estão para morrer. Embora ilegal e inapropriado, a clonagem humana é feita clandestinamente, existindo uma empresa americana, a ClonaidTM, especializada nesta técnica. Embora formalmente não exista, a empresa, já com alguns anos, dispõe de um site na internet que permite aos clientes contactar e requisitar os serviços, que nunca foi fechado pelas autoridades. O diretor é conhecido, sendo ainda de conhecimento público que já se “nasceu” a primeira criança clonada, mas a empresa recusa-se a revelar o paradeiro da mesma, bem como de todos os outros clones que alegadamente foram criados nos últimos 6 anos. A empresa já foi a tribunal. BRAINER MOUSE Já se modificou ratinhos nomeadamente a nível dos genes codificantes de recetores neurais (NMDA) que promovem a ligação entre células neurais, criando: Dumb mouse: fez-se o knockout do gene NMDA e obteve-se um ratinho com maior dificuldade em fazer ligação entre células neurais e pouca memória. Este apresentava uma memória espacial prejudicada, não recordando os caminhos anteriormente feitos até à comida. Doogie mouse: adicionaram-se cópias extras do gene NMDA e observou-se um maior número de conexões entre as células neurais e uma capacidade de memória 4 a 5x superior aos ratinhos normais. Há que ter em conta que também a engenharia genética tem limitações, citando que “nunca irá transformar um rato num génio capaz de tocar piano”. – Scientific American, Abril 2000.
Mais conteúdos dessa disciplina
 02-Autophagosome-based strategy to monitor apparent tumor-specific CD8 T cells in patients with p
02-Autophagosome-based strategy to monitor apparent tumor-specific CD8 T cells in patients with p- 07-RETRACTED ARTICLE_ Rational design of multifunctional micelles against doxorubicin-sensitive a
- 07-RETRACTED ARTICLE_ Combination therapy with Nab-paclitaxel and the interleukin-15 fused with a
- 06-RETRACTED ARTICLE_ The role of calgranulin B gene on the biological behavior of squamous cervi
- 05-RETRACTED ARTICLE_ microRNA-605 inhibits the oncogenicity of non-small-cell lung cancer by dir
- 07-RETRACTED ARTICLE_ KLF7 Promotes Gastric Carcinogenesis Through Regulation of ANTXR1
- 06-RETRACTED ARTICLE_ KLF7 Promotes Gastric Carcinogenesis Through Regulation of ANTXR1
- 05-RETRACTED ARTICLE_ MicroRNA-155-3p promotes breast cancer progression through down-regulating
- 04-RETRACTED ARTICLE_ miR-489-3p Inhibits Prostate Cancer Progression by Targeting DLX1
- 02-RETRACTED ARTICLE_ Overexpression and clinical significance of MYC-associated zinc finger prot
- 00-Pigment epithelial-derived factor gene loaded novel COOH-PEG-PLGA-COOH nanoparticles promoted
- Na década de 1960 um artigo acadêmico foi publicado por Theodore Levitt. Esse artigo, intitulado Miopia de Marketing, defendia que: As empresas já ...
- 182
- 183
Conteúdos escolhidos para você





Perguntas dessa disciplina
Mais conteúdos dessa disciplina
- 02-Autophagosome-based strategy to monitor apparent tumor-specific CD8 T cells in patients with p
- 07-RETRACTED ARTICLE_ Rational design of multifunctional micelles against doxorubicin-sensitive a
- 07-RETRACTED ARTICLE_ Combination therapy with Nab-paclitaxel and the interleukin-15 fused with a
- 06-RETRACTED ARTICLE_ The role of calgranulin B gene on the biological behavior of squamous cervi
- 05-RETRACTED ARTICLE_ microRNA-605 inhibits the oncogenicity of non-small-cell lung cancer by dir
- 07-RETRACTED ARTICLE_ KLF7 Promotes Gastric Carcinogenesis Through Regulation of ANTXR1
- 06-RETRACTED ARTICLE_ KLF7 Promotes Gastric Carcinogenesis Through Regulation of ANTXR1
- 05-RETRACTED ARTICLE_ MicroRNA-155-3p promotes breast cancer progression through down-regulating
- 04-RETRACTED ARTICLE_ miR-489-3p Inhibits Prostate Cancer Progression by Targeting DLX1
- 02-RETRACTED ARTICLE_ Overexpression and clinical significance of MYC-associated zinc finger prot
- 00-Pigment epithelial-derived factor gene loaded novel COOH-PEG-PLGA-COOH nanoparticles promoted
- Na década de 1960 um artigo acadêmico foi publicado por Theodore Levitt. Esse artigo, intitulado Miopia de Marketing, defendia que: As empresas já ...
- 182
- 183